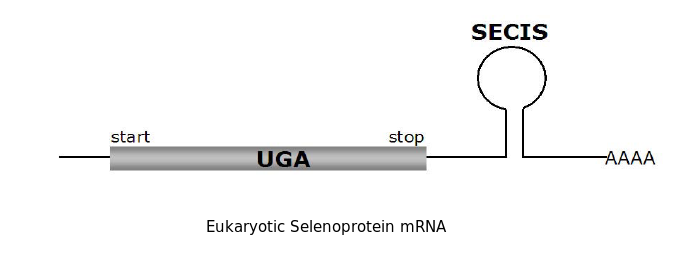
Selenoproteins
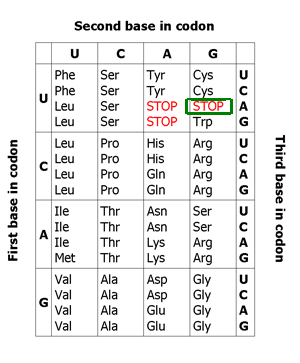
Selenoproteins are proteins that contain selenocysteine, an aminoacid that is analogous to cystein but that has selenium (an essential micronutrient in the diet) in spite of sulphur. These selenoproteins need an specific machinery of synthesis that is conserved in the different species in which selenoproteins have been described. In selenoproteins, the incorporation of the selenocystein aminoacid is specified by the UGA codon, that commonly codifies for a stop signal (genetic code). The alternative decodification of this codon is performed thanks to an mRNA structure: the SECIS element, that can be found in UTR 3'. The SECIS elements are defined by a characteristic sequence and by an specific secondary structure that is formed because of the basepairy, this fact represents a non canonic basepairing (U-G) in eucariotes. Because the major part of prediction programs interpret UGA codon as a stop codon, these programs ara unable to identify selenoproteins which makes it necessary to work out other bioinformatic methods to detect the presence of a selenoprotein.
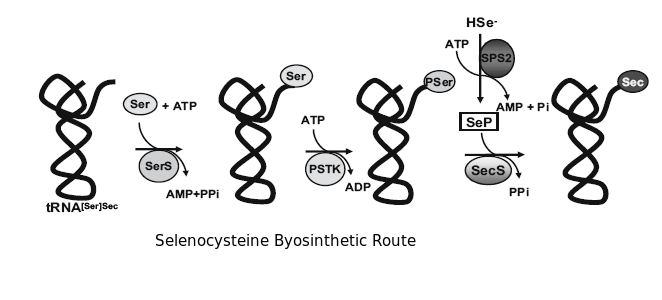
Selenocystein synthesis
Selenocystein synthesis comprises the following steps:
- Serine tRNA aminoacylation with a seril-tRNA synthetase.
- Seril-tRNA synthetase phosphorylation by PSTK (PSTKinase), allowing the formation of a O-phosphoseril tRNA.
- Dephosphorilation of the tRNA that produces the acceptor molecule for SeP.
- SeP previous synthesis by SPS2, so SeP is responsible for the last step to obtain selenocystein.
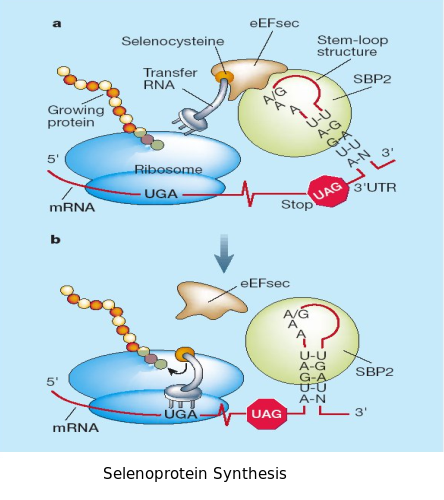
SECIS element
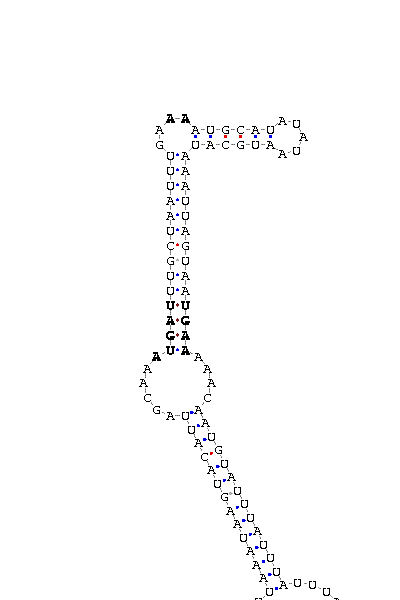
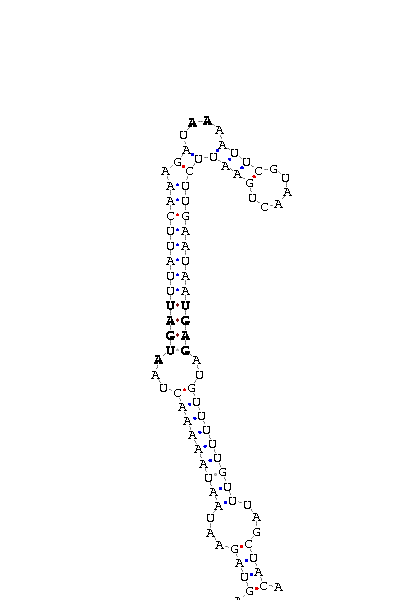
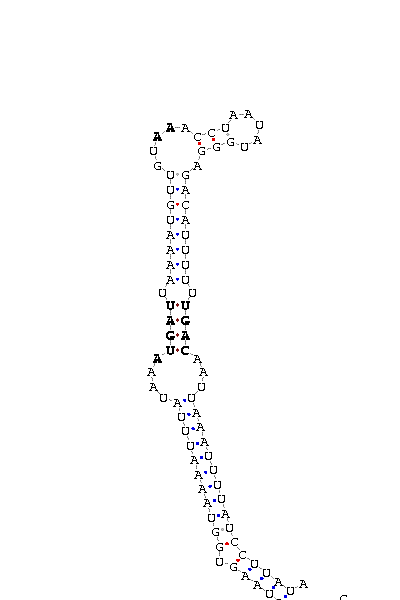
As we have already explained, the SECIS elements are stem-loop structure sequences located in the 3'UTR end, that are needed for ignoring the STOP codon and being able to include in this position a selenocysteine. The SECIS element is located after the UGA codon, and sometimes even some Kb after it. Its secondary structure has been fairly maintained, and contains consensus characteristic sequences, as we can here appreciate in the red color of this image.
Function of Selenoproteins
The presence of such a complex and specific machinery makes us think that the function of these proteins is not trivial. Actually, these proteins are involved in catabolic processes and in the redox reactions catalization. In these last reactions they produce an antioxidant effect, avoiding harmful effects that cause several alterations. Every Selenoprotein has a protective effect against different free radicals (Hidrogen peroxide, Phospholipid hidroperoxides, etc.)
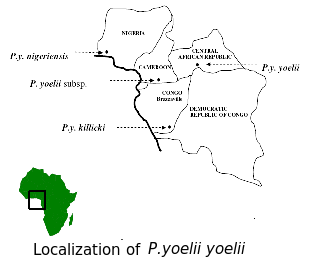
Plasmodium yoelii yoelii
Plasmodium yoelii yoelii is an eukaryotic organism (kingdom Alveolata, filum Apicomplexa, class Aconoidasida, order Haemosporida) that has been isolated from blood of shiny thicket rats (Thamnonys rutilans from Central Republic of Africa, Brasil and the west part of Nigeria). Three P.yoelii subspecies can be distinguished:P. yoelii yoelii, P. yoelii killicki and P. yoelii nigeriensis.
These organisms are parasites that cause the malaria disease in rodents. Because of that, Plasmodium yoelii yoelii (and others) are used spreadly as a model in functional analyses in order to identify new medicines and targets to treat and prevent this disease (it is grown in the laboratory in rats and mice; and it has been demonstrated that it prefers inmature erythrocytes and retyculocytes).
Malaria disease is an infeccious disease caused by the Plasmodium genus and that it is caught by a vector, the female Anopheles mosquito. Every year, this illness causes 515 millions of sick people and kills between 1 and 3 millions of people, the majority of whom are children from Sub-saharan Africa. Therefore, the malaria represents an important problem in public health.
Inside the Plasmodium genus just four species infect mankind:Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale i Plasmodium malariae. Other species are able to infect other organisms, as Plasmodium yoelii.
Malaria's cycle
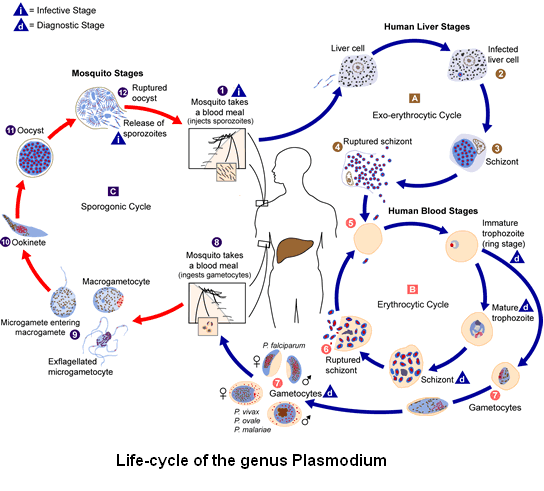
The malarial parasite's life-cycle includes two hosts: the female Anopheles mosquito and human.
When a female Anopheles infected with Plasmodium feeds with human blood, it inoculates sporozoytes (1). The sporozoytes infect the hepatocytes (2) and mature to schizont (3), who break the cells and release the merozoytes (4)(in P.vivax and P.ovale there is a latent stage [hypnozoite] that can persist in the liver and cause relapses due to its release to the bloodstream weeks or years later). After this initial replication in the liver (Exo-erythrocytic cycle [A]), the parasite reproduce themselves in an asexual way in the erythrocytes (Erythrocytic cycle [B]). The merozoytes infect the erythrocytes (5). The trophozoyte stage mature to schizont, who cause the cellular breakage that leads to merozoytes (6). Some parasites differ in the sexual erythrocytic stages (7). The stages occuring in blood are the cause of the classical signs of the malaria desease.
The male gametocytes (microgamonts) and female (macrogamonts) are eaten by the female Anopheles mosquito during one of its alimentation periods (8). The multiplication of parasites inside the mosquito is known as the Sporognic cycle [C]. In the meantime, in the mosquito's stomach the microgamonts penetrate the microgamonts, producing zygotes (9). The zygotes become movile and extended (known as oocinets) (10), who invade the mosquito's intestinal wall where they develope to oocysts (11). The oocysts grow and turn into sporozoytes (12), who move to the mosquito's salivate glands. The inoculation of sporozoytes to a new human host perpetuates the malaria cycle (1).
Tot i que s'estan desenvolupant algunes vacunes contra la malària, el tractament bàsic es basa en fàrmacs profilàctics dels quals cada vegada és més comú detectar-ne resistències. Although some vaccines against malaria are nowadays under development, the basic treatment is based in prophylactic medicines. However, resistence has been detected against these medicines. That is why the Plasmodium species that affect humans are being deeply studied in parallel to species that affect laboratory animals, like Plasmodium yoleii. They allow us to investigate in order to reach a satisfactory treatment for this disease.
The Institute for Genomic Research (TIGR) in cooperation with the Naval Medical Research Center have concluded a research programm to sequenciate the complete genome of Plasmodium falciparum, Plasmodium vivax, P. yoelii, P. berghei and P. chabaudi, that provide us with a deep knowledge of the evolutive history of these species. The specific data from the P. yoelii yoelii genome used in this work can be found in the "Materials & Methods" section.
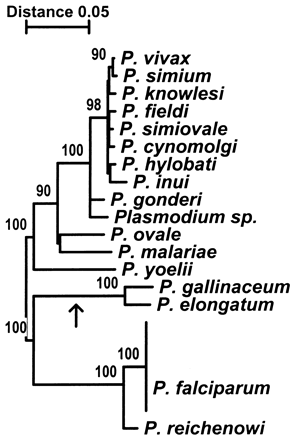
Phylogenetic tree image: Phylogenetic relationship among the 17 Plasmodium species inferred from the gene encoding cytochrome b. The tree was estimated using the NJ method.Image extracted from http://www.pnas.org/cgi/content/full/95/14/8124?ck=nck
Research of Plasmodium yoelii yoelii's genome and other organisms selenoproteins
First of all, the genome of Plasmodium yoelii yoelii (str 17XNL) that has been obtained, was provided by the project supervisors. However, it can be found at plasmodb.org and NCBI.
Plasmodium yoelii yoelii genome characteristics
The project to sequence the whole genome (Whole Genome Shotgun) consists in an anotation that has been genered authomatically, it is preliminar and is formed by contigs. Consequently, it is submitted for revision because it is necessary to go deeper in the anotation in order to finish it. The contigs represent more than 2kb (20Mb in total), geting an 87% of the whole genomic sequence. The genetic code of Plasmodium yoelii yoelii's genome belongs to table 1 (standard) while the mithocondrial genetic code belonts to translation table 4.
On the other side, sequences of selenoproteins and proteins that belong the their synthesis machinery of other organisms have been obtained thanks to different sources:
- Selenodb database: sequences of Homo sapiens, Pan troglodytes, Mus musculus, Tetraodon nigroviridis, Drosophila melanogaster, Anopheles gambiae, Caenorhabditis elegans and Saccharomyces cerevisae. A multifasta document has been generated for each organisme that shows all its proteins.
- NCBI database: Plasmodium falciparum and Plasmodium yoelii sequences, as well as Drosophila melanogaster protein sequences. The sequences have been obtained from this application, since we have obtained the access codes for these proteins from scientific articles quotated at the references.
Using BLAST to research similarity
The sequences of the selenoproteins and proteins of the synthesis machinery from different organisms have been compared with the Plasmodium yoelii yoelii genome. In this case, TBLASTN has been used because it allows to compare aminoacidic sequences of proteins (query) with nucleotide sequences belonging to the Plasmodium yoelii yoelii genome (subject) due to its capacity of translating the nucleotide sequence in each of the possible open reading frames.
The alignments with TBLASTN have been performed using the command prompt. Previously the genome of Plasmodium yoelii yoelii has been given format with:
formatdb -i genome.fa -p F
-p F indicates a nucleotidic genome.
After having installed BLAST, it has been used the command:
blastall -p tblastn -d database -i query.fa
Where database belongs to the Plasmodium yoelii yoelii genome and the query.fa belongs to the aminoacidic sequence
The remarkable parameters of TBLASTN configuration choosed to perform the alignmetns are:
- BLOSUM62 matrix= substitution matrix based in local alignments. The number corresponds to the identity of the sequences used to build up the matrix. The similarity percentage is not very high due to the long evolutive distance that can be found between Plasmodium yoelii yoelii and the organisms with which it is compared.
- e-value = 10.0. This value is very high, taking into account that ideally it should be 0.1. Another time, it is important to remark that the alignment has been done between proteins of organisms evolutively far from Plasmodium yoelii yoelii. However, the e-values superior to 1 haven't been analyzed.
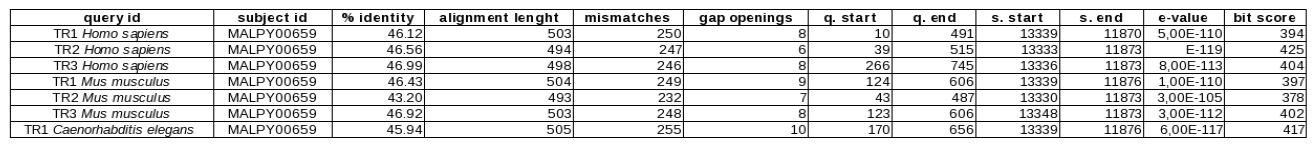
- -m 9 format= table style format that shows accurate and classified information of the most representative alignments in different aspects: query identity, subject identity, % identity, alignment length, mismatches, gap openings, query start, query end, subject start, subject end, e-value and bit score.
The identification of the most outstanding matches is achieved using TBLASTN. Thus, what has been really done, is to restrict the genome areas that are potential selenoproteins genes, selenoprotein machinery or proteins homologous to selenoproteins. In the case of selenoproteins the objective is to identify the alignments where the selenocystein aminoacid is aligned with a genome's stop codon. Contrary, in the case of homologous proteins it is interesting to find the alignments that coincide with a cysteine. Referring to the machinery proteins, it depends on whether they are selenoproteins or not: in the first case, they are analyzed as selenoproteins, in the second case as homologous proteins, without taking into account any specific aminoacid.
Obtaining the genomic sequence of interest
In order to improve the posterior analysis, the previous aligned sequence has been cut off from its contig. This procedure has been done as follows:
perl FastaToTbl.pl < genome.fa | awk '$1=="subject_identity"' | perl TblToFasta.pl output.fa
fastasubseq -f (fasta file) -s (inici) -l (llargada) > output.fa
With the first command and using two perl programmes, the contig of interest is isolated creating a new fasta document. The second command cuts the specific sequence of nucleotides inside the contig.
Searching similarity and structure: Exonerate and GeneWise
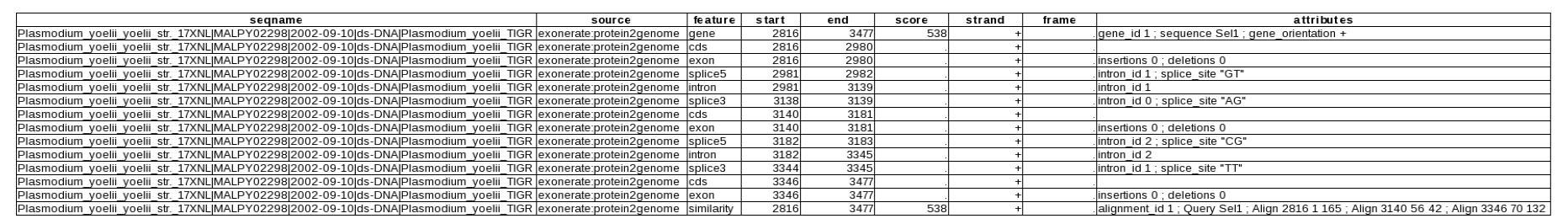
The following step is to predict the gen structure. Exonerate and GeneWise are programs that allow the prediction of the exonic structure taking into account the presence of introns. Both programs realign the two sequences; having restricted the nucleotide sequence of interest, the alignment conditions are improved. Exonerate has been used as follows:
export PATH=$PATH:/disc8/bin/exonerate/bin
exonerate -m p2g -q query.fa -t target.fa --showtargetgff
query.fa corresponds to the aminoacidic sequence of an organism protein and target.fa to the previous chosen genomic region of Plasmodium yoelii yoelii. The gff format allows to summarize in a table the most important information about the gene. GeneWise has been used from its web page www.ebi.ac.uk with the Advanced GeneWise option, choosing the following: gene structure, translation, cDNA, GFF output, global alignment and organism worm.
Research of SECIS elements: SECISearch
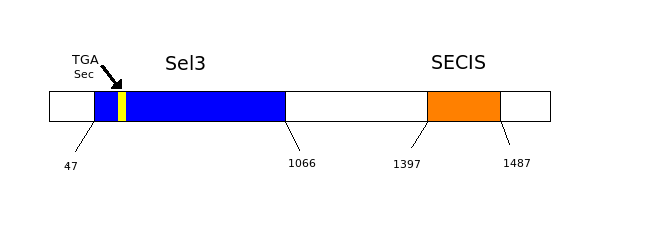
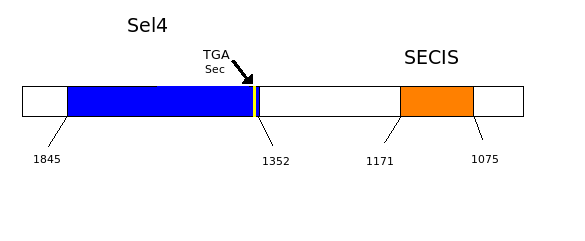
When the gene codifies for a selenoprotein, the SECISearch program has been used to search for the potential SECIS element at 3'UTR. It is important to take into account that it is necessary to search for the SECIS element at a minimum distance of 3000bp downstream, because up to the moment it is reported that the SECIS element is separated from the codifying region of the gene.
Translating genes: Expasy
Given the exonic sequences predicted by Exonerate o GeneWise, they have been translated in order to obtain the predicted sequence for each single gene. This process is achieved with the Translate application from the Expasy server that translates the sequence into the six possible frames (three for each sense) and chosses the best one.
Aligning sequences: ClustalW
The sequences of Plasmodium yoelii yoelii proteins and the protein sequences of the organisms used in each case have been aligned by ClustalW, making it possible to show their similarities and differences. With this last step the conservation between both sequences can be observed.
The web sources used are:
National Center for Biotechnology Information
Plasmo database
Seleno database
GeneWise
SECISearch
ExPaSy Proteomics Server
ClustalW
Pfam Home Page
Note
This is a general protocol, that is to say, the procedures have been explained at a global level. However, it is important to take into account that each gene has been a different challenge and from the first alignment to the obtention of the final protein in some cases it has been difficult to solve the problems only by following the general protocol.
Because of that, each gene has an specific section in which the specific procedure is explained.
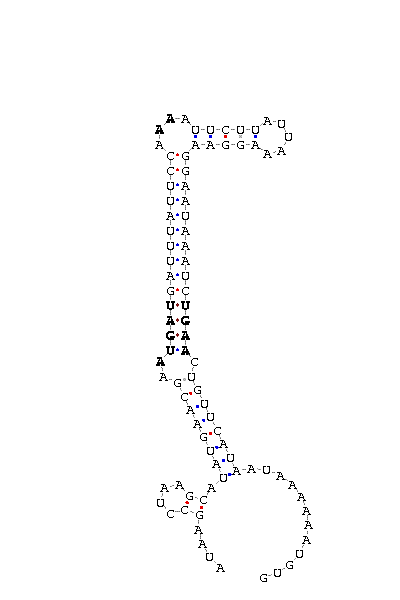
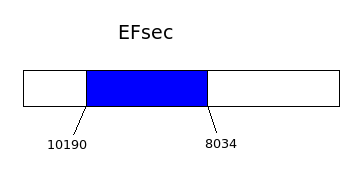

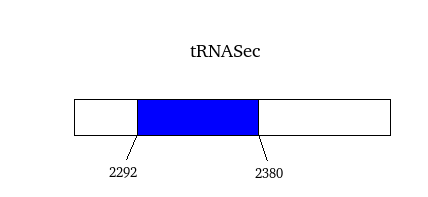
.png)
.png)
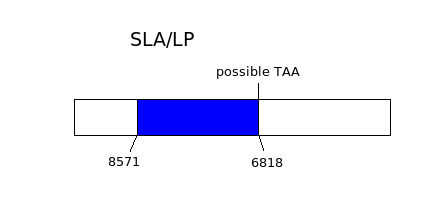
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
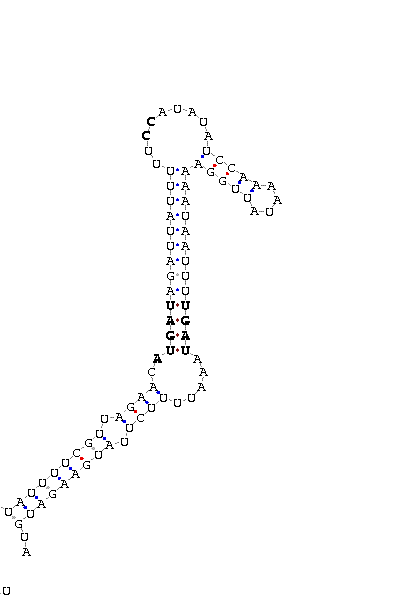{kind=link}
{kind=link}
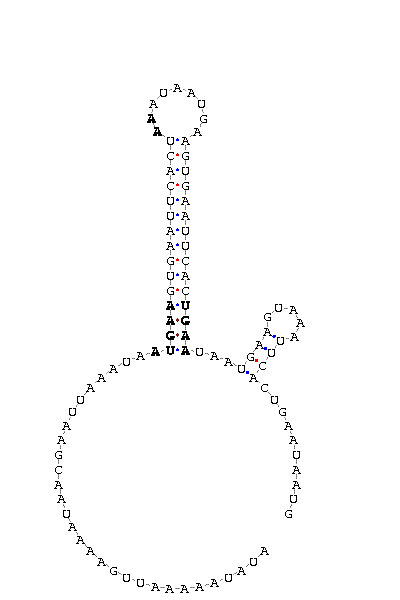{kind=link}