Anàlisi del cluster HOX A
Núria Ramos González
Mar Mallo Fajula
Facultat de Ciències de la Vida i la Salut, UPF
Aquest treball es basa en l'anàlisi exhaustiu de la regió del cluster HOX A del genoma, localitzat en el cromosoma 7p i de 0.5 MB.
Aquest anàlisi pretén simular en certa manera un projecte d'extraordinària importància, el projecte ENCODE.
El projecte ENCODE (Encyclopedia Of DNA Elements) és un consorci de recerca públic que nasqué l'any 2003 amb l'objectiu d'identificar tots els elements funcionals en el genoma humà. Aquest projecte implica l'interacció entre científics experimentals i computacionals per tal d'avaluar diversos mètodes per establir les anotacions del genoma humà. S'ha seleccionat un conjunt de regions que representen aproximadament un 1% (30 MB) del genoma les quals estan éssent analitzades. Tota la informació generada és dipositada en bases de dades púpliques, de manera que l' ENCODE Consortium és obert a tots els sectors.
Com hem dit el nostre treball s'ha basat en l'estudi de la regió que engloba el cluster HOXA, per això, farem una breu introducció als gens Hox.
La família de gens hox codifiquen per a un grup de factors de transcripció responsables de regular la morfogènesi i de conferir la identitat axial en el desenvolupament de l'embrió durant la gastrulació. Els gens Hox tenen en comú una seqüència de DNA coneguda com a caixa homeòtica (homeobox)present en diferents organismes tant invertebrats com vertebrats.
El complex gènic en Drosophila es localitza en el cromosoma 3 i conté els clusters Antennapedia i Bithorax.
Els vertebrats tenen 39 gens hox i el complex homeòtic comprèn quatre clusters organitzats en 13 grups paràlegs. Aquests clusters (gens que són vistos com a una unitat funcional) s'anomenen HOXA, HOXB, HOXC I HOXD i donada la gran homologia entre els gens hox de Drosophila i els vertebrats, es pensa que els gens Hox de vertebrats són fruit de la duplicació cromosòmica dels gens de Drosophila. Així doncs, l'homologia en l'estructura gènica i la similaritat en el patró d'expressió entre Drosophila i els gens Hox de mamífer suggereix que aquest mecanisme de patterning és extremadament ancestral.
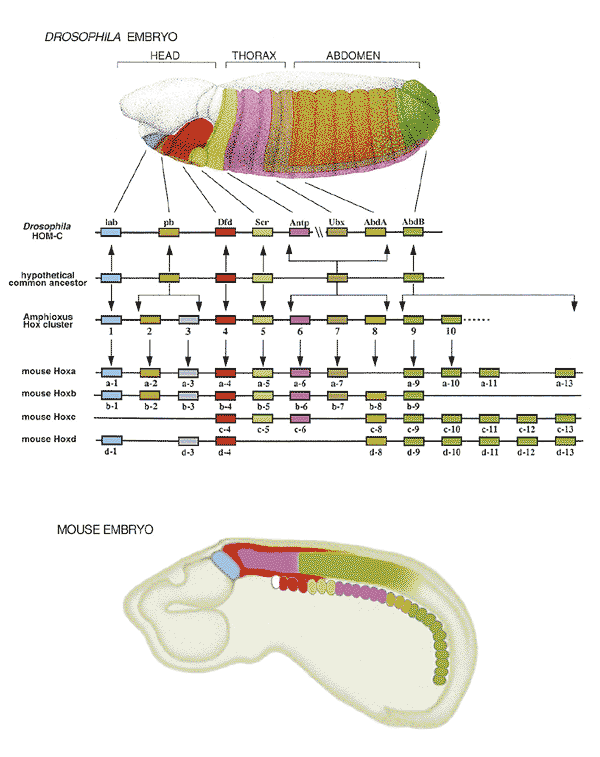
Figura 1. Homologia entre gens hox de Drosophila i de ratolí
La funció dels gens Hox consisteix a establir el patterning de l'eix antero posterior tot i especificant les posicions al llarg d'aquest eix. L'organització cromosòmica dels gens Hox de cada cluster reflexa l'expressió antero posterior en els eixos dels cos, és a dir, presenten colinearitat espacial (expressió colinear de 3' a 5'). Així doncs, aquests gens s'expressen al llarg de l'eix dorsal des del llindar del hindbrain fins la cua (tub neural, cresta neural, mesoderm paraaxial i superfície de l'ectoderm).
A més a més l'expressió diferencial de gens hox especifica les diferents regions al llarg de l'eix, així doncs, les combinacions de diferents gens especifiquen una regió particular al llarg de l'eix antero-posterior. Diferents experiments ens avalen la seva funció. Al realitzar un KO dels gens Hox, s'esdevenen malformacions específiques de segment, d'altra banda l'expressió ectòpica d'aquests gens altera l'establiment dels eixos corporals.
Per tal de portar a terme l'anàlisi de la seqüència vam començar determinant la seva localització en el genoma, la seva longitud i la proporció de cadascun dels nucleòtids.
Posteriorment, vam emmascarar la seqüència mitjançant el servidor web del RepeatMasker. D'aquesta manera, aconseguim determinar els elements repetitius presents en la nostra seqüència.
Un dels objectius del treball és la predicció dels gens que podem trobar en la nostra seqüència, ho aconseguim mitjançant tres programes que funcionen via servidors web: el GENSCAN, el GENEid i el FGENESH.
Cadascun dels programes de predicció de gens donen resultats similars, tot i que no idèntics, per fer-ne una selecció ens basem, bàsicament, en la validació a partir dels ESTs.
Un cop tenim els gens seleccionats, caracteritzem les proteïnes predites, mitjançant la cerca en bases de dades de proteïnes humanes (BLASTP).
Aquest treball s'ha realitzat a travé de diversos servidors web emprant el sistema operatiu LINUX.
1) Obtenció de la seqüència:
2) Emmascarament de la seqüència:
3) Predicció de gens "ab initio":
4) Validació de la predicció de gens
5) Aliniament de proteïnes
6) Dominis proteics:
7) Programes
gff2ps
Parseblast
Fastachunk
Formatdb
Kview
*Cal recordar que per fer servir els diversos programes cal fer previs exports:
export PATH=$PATH:/disc8/bin
export PATH=/disc8/soft/perl/bin/:/disc8/bin/:$PATH
export PATH=$PATH:/disc8/bin/:/disc8/bin/ncbiblast/:/disc8/soft/R/bin
export BLASTMAT="/disc8/bin/ncbiblast/data"
Per tal d'analitzar la seqüència procedim a descarregar-la del UCSC Genome Browser on Human July 2003 del Projecte ENCODE.

Figura 2. Localització de la seqüència.
- Identificador de la regió analitzada: ENm010.
- Posició: 26,699,793-27,199,792.
- Tamany: 0,5 MB.
Guardem la seqüència localment com a: hox.fa.
Per tal de realitzar l'anàlisi preliminar cal tabular la seqüència obtinguda en FASTA mitjançant la següent comanda:
awk '{printf $1}' hox.fa > hox.tbl0
Amb l'emacs introduim un tab ("\t") entre el nom de la seqüència i la seqüència en si, per tal d'aconseguir el format tabulat adequat. Aquest fitxer l'anomenem hox.tbl.
Amb les següents comandes obtenim:
- La longitud de la seqüència: 500.000pb
awk '{print length($2)}' hox.tbl
awk '{print $2}' hox.tbl | fold -1 | sort | uniq -c | gawk '{print $2, $1/500000}'
Com ja sabem el genoma humà presenta un elevat percentatge de regions repetitives (que pertanyen a diferents famílies) les quals poden interferir en aquest anàlisi.
Per tant, cal emmascarar aquestes regions de la seqüència mitjançant un programa que compara la seqüència problema amb una base de dades d'elements repetitius. Nosaltres emprarem el servidor EMBL del Repeat Masker.
Introduim la seqüència hox.fa i activem les opcions:
- Running options:"fast" que tot i que té menor sensibilitat el procés és més ràpid.
El Repeat Masker dona com a resultat diversos outputs:
- hox.seq.out (annotacions de la seqüència emmascarada)
Resum del contingut de repeticions:
- Bases emmascarades: 174803 pb
Observem que la major part de repeticions corresponen a SINEs i LINEs i que es troben bàsicament en la mateixa proporció tot i que n'hi ha d'altres tipus.
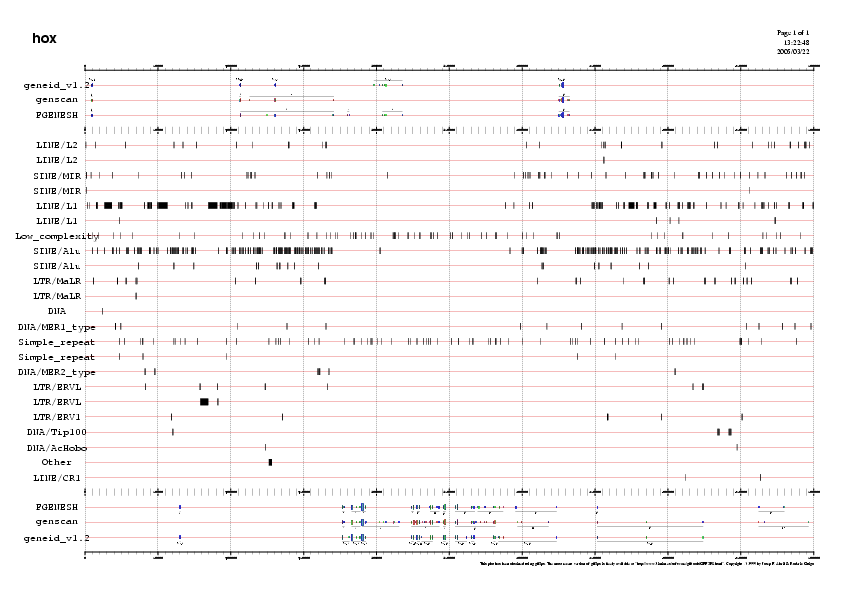
En aquest punt visualitzarem com es distribueixen aquestes repeticions al llarg del genoma:
1) convertir el fitxer hox.seq.out a gff mitjançant la comanda:
grep hox hox.seq.out | \
awk ' BEGIN{ OFS= "\t" }
{print $5, $11, "repeat", $6, $7, ".", ".", "."; }
' > hox.seq.out.gff
2) Obtenim el post script amb el programa gff2ps:
gff2ps hox.seq.out.gff > hox.seq.out.ps
3) Convertim el fitxer obtingut a png per tal de visualitzar-ho amb el Kview:
convert -antialias -rotate 90 hox.seq.out.ps hox.seq.out.png
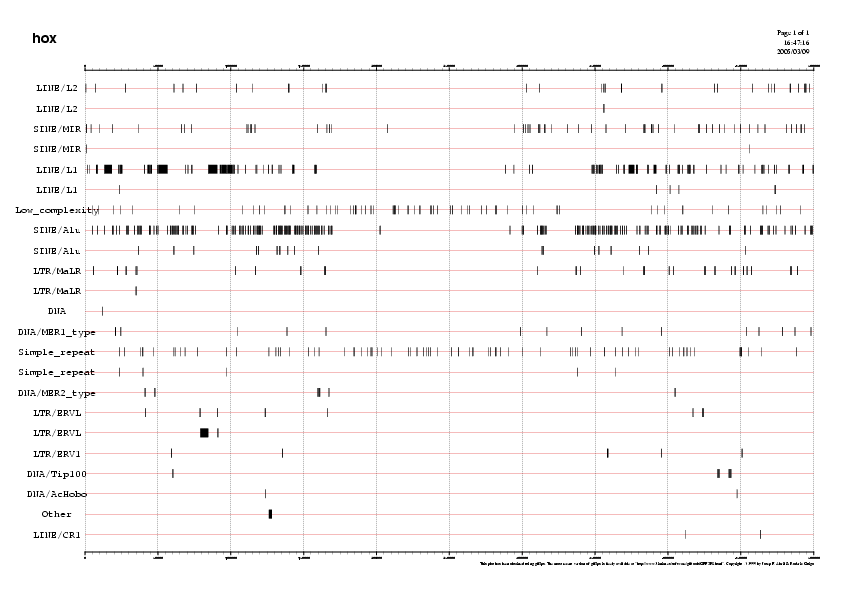
Figura 3. Distribució dels elements repetitius
La predicció dels gens presents en la nostra seqüència genòmica van ser predits a partir de tres programes de predicció de gens: GENSCAN, GENEid i FGENESH.
1. Predicció per GENEid:
Introduïm la seqüència emmascarada en el browser i seleccionem les següents opcions de predicció:
- Organism: Homo sapiens.
- Prediction models: Normal mode.
- DNA strands: Forward and Reverse.
El GENEid ens permet obtenir dos tipus d'outputs, un en format GENEid (hoxmasked.geneid.txt) i, un altre, en format gff (hoxmasked.geneid.gff).
2. Predicció per GENSCAN:
El programa GENSCAN ens dona com a output un fitxer txt (hoxmasked.genscan.txt) que cal transformar a gff (hoxmasked.genscan.gff):
gawk 'BEGIN{OFS="\t"}
$2 ~ /Term|Intr|Init/ {
print "hox", "genscan", $2, start=($4<$5 ? $4 : $5),
end=($5<$4 ? $4 : $5), $13, $3, $7, $1;
}' hoxmasked.genscan.txt | \
sed 's/\.[0-9][0-9]$//' > hoxmasked.genscan.gff
3. Predicció per FGENESH:
Obtenim un fitxer en format txt (hoxmasked.fgenesh.txt) i el transformem a format gff (hoxmasked.fgenesh.gff), a través d'un programa que ens permeti posar el frame i el cridem de la següent manera:
./fgenesh2gff.awk "hox" hoxmasked.fgenesh.txt > hoxmasked.fgenesh.gff
4. Gens resultants de les prediccions:
| Gen | GENEid | Gen | GENSCAN | Gen | FGENESH |
| 1 |
4260-5011 (+) |
1 |
4260-5011 (+) |
1 |
4260-5011 (+) |
| 2 |
65008-65430 (-) |
3 |
105837-107008 (+) |
2 |
65008-65430 (-) |
| 3 |
105837-105935 (+) |
4 |
112818-170728 (+) |
3 |
106574-170728 (+) |
| 4 |
106574-107008 (+) |
5 |
176539-177996 (-) |
4 |
176539-177996 (+) |
| 5 |
130268-130602 (+) |
6 |
182825-189000 (-) |
5 |
180044-181284 (+) |
| 6 |
176539-181365 (-) |
7 |
190014-215505 (-) |
6 |
182825-189000 (-) |
| 7 |
182825-188952 (-) |
8 |
223934-243225 (-) |
7 |
190014-192739 (-) |
| 8 |
190014-192739 (-) |
9 |
245702-247556 (-) |
8 |
204074-217751 (+) |
| 9 |
198913-217751 (+) |
10 |
253998-281627 (-) |
9 |
223934-225589 (-) |
| 10 |
223934-225589 (-) |
11 |
296642-318103 (-) |
10 |
227757-229848 (-) |
| 11 |
227757-229848 (-) |
12 |
325130-332644 (+) |
11 |
236698-243225 (-) |
| 12 |
230855-238644 (-) |
13 |
351431-424114 (-) |
12 |
245702-247556 (-) |
| 13 |
245702-247556 (-) |
14 |
462161-496616 (-) |
13 |
253998-267579 (-) |
| 14 |
253998-262009 (-) |
- |
------ |
14 |
269414-286985 (-) |
| 15 |
264895-267117 (-) |
- |
------ |
15 |
295308-323701 (-) |
| 16 |
280297-281546 (-) |
- |
------ |
16 |
325130-332385 (+) |
| 17 |
283588-323701 (-) |
- |
------ |
17 |
351431-351810 (-) |
| 18 |
325400-328524 (+) |
- |
------ |
18 |
462161-479933 (-) |
| 19 |
351431-424150 (-) |
- |
------ |
- |
------ |
Taula 1. Predicció de gens
NOTA: Resaltem aquells gens que són igualment predits pels tres programes.
5. Visualització de les prediccions:
- Convertir els outputs .gff a ps mitjançant el gff2ps i obtenim els fitxers:
hoxmasked.geneid.ps
hoxmasked.genscan.ps
hoxmasked.fgenesh.ps
- Posteriorment convertim els fitxers .ps a .png per tal de visualitzar-los amb el Kview:
convert -antialias -rotate 90 hoxmasked.geneid.ps hoxmasked.geneid.png
convert -antialias -rotate 90 hoxmasked.genscan.ps hoxmasked.genscan.png
convert -antialias -rotate 90 hoxmasked.fgenesh.ps hoxmasked.fgenesh.png
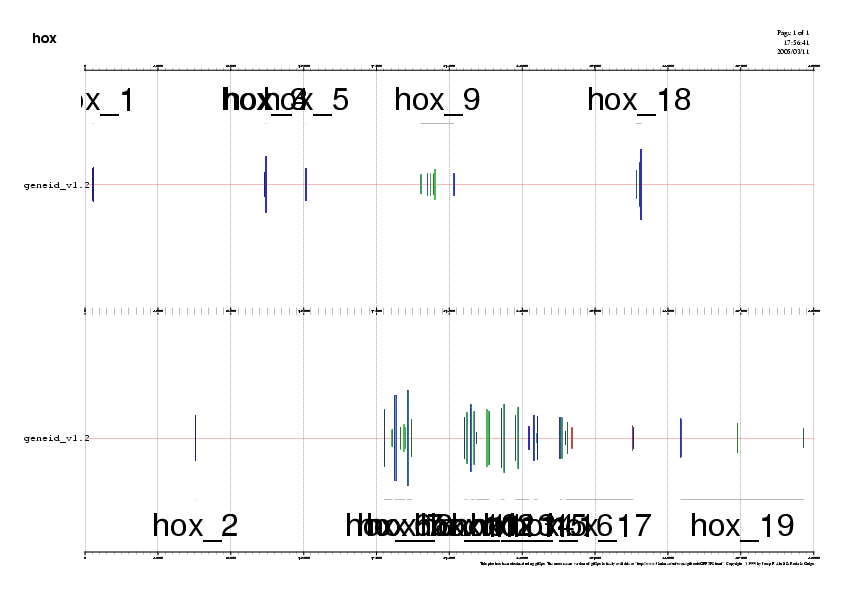
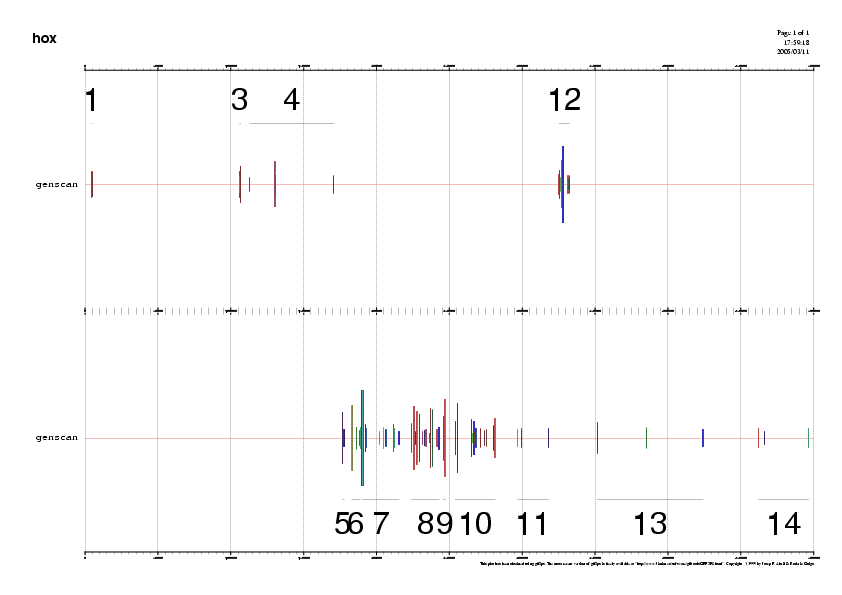
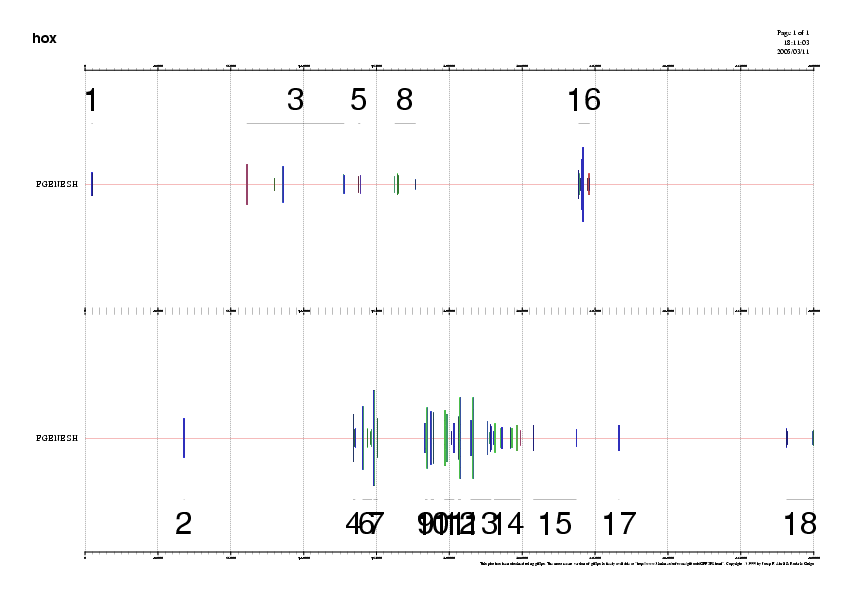
Figura 4. Predicció del GENEid Figura 5. Predicció del GENSCAN Figura 6. Predicció del FGENESH
- Per tal de poder extreure conclusions cal visualitzar les tres prediccions de manera simultània
gff2ps hoxmasked.geneid.gff hoxmaskedp.genscan.gff hoxmasked.fgenesh.gff > hox.genepredictions.ps
- Posteriorment cal convertir el fitxer .ps a .png per tal de veure'l amb el Kview
convert -antialias -rotate 90 hox.genepredictions.ps hox.genepredictions.png
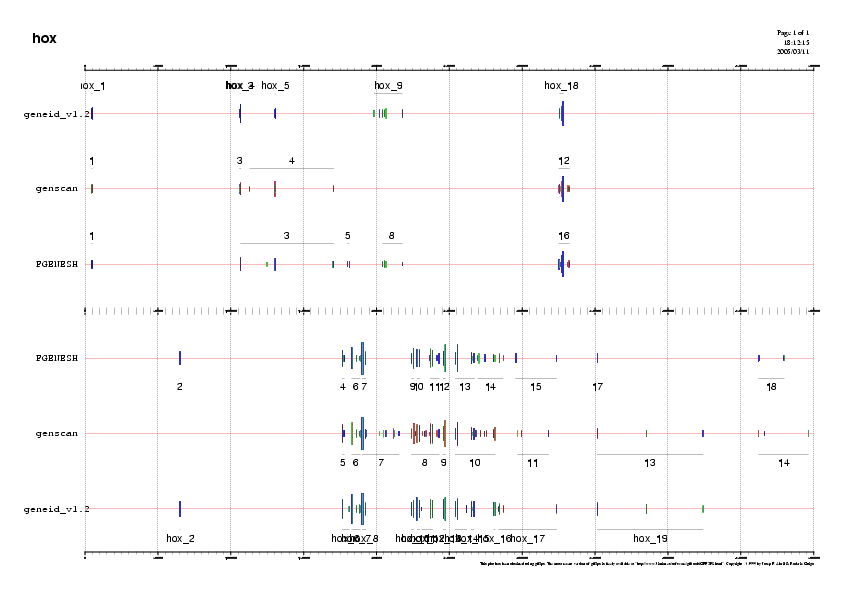
Figura 7. Gens predits pels tres programes
També podem fer servir la següent comanda per tal de poder visualitzar més acuradament les prediccions, mostrant 5 blocs distribuïts en quatre pàgines:
gff2ps -B 5 -P 4 hoxmasked.geneid.gff hoxmasked.genscan.gff hoxmasked.fgenesh.gff > hox.genepredictions_B5P4.ps
Posteriorment es converteix el post script a png i es visualitza amb el Kview.
convert -antialias -rotate 90 hox.genepredictions_B5P4.ps hox.genepredictions_B5P4.png
hox.genepredictions_B5P4.png.0
hox.genepredictions_B5P4.png.1
hox.genepredictions_B5P4.png.2
hox.genepredictions_B5P4.png.3
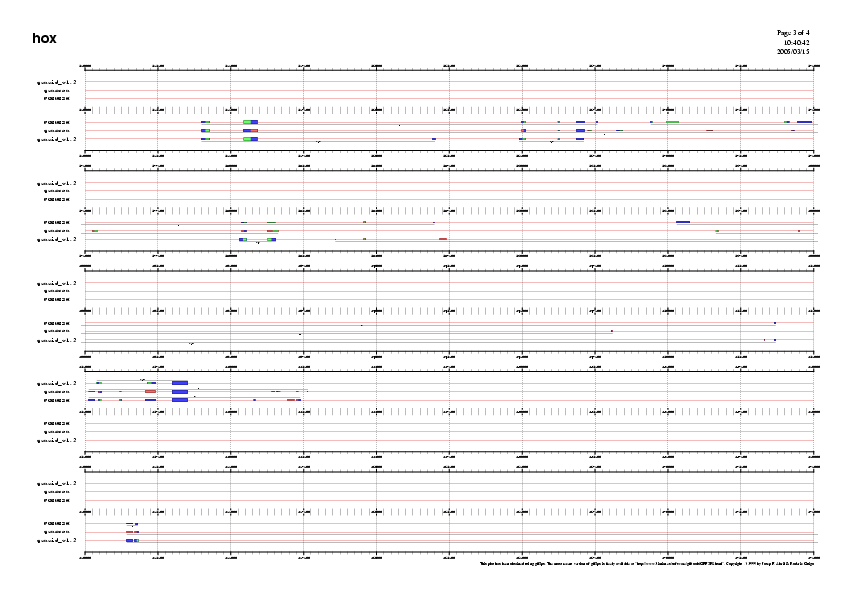
Figura 8-11. Ampliació dels gens predits pels tres programes
A partir dels resultats obtinguts observem que si bé els diferents programes predeixen gens similars, les prediccions no són exactes en tots els programes.
El GENSCAN ha predit 14 gens, el FGENESH n'ha predit 18 i el GENEid 19.
Observant el gràfic veiem que la regió central de la seqüència presenta alta densitat de gens, mentre que d'altres zones presenten un contingut gènic menor.
A més a més, el número de gens predit en reverse és superior al número predit en forward.
Per últim, es realitza un gràfic que mostra de manera conjunta els elements repetitius de la seqüència i les prediccions gèniques pels diferents programes.
gff2ps hoxmasked.geneid.gff hoxmasked.genscan.gff hoxmasked.fgenesh.gff hox.seq.out.gff > repgens.ps
I per visualitzar-ho amb el Kview, ho passem a format png (repgens.png)
Observem que les regions amb altes densitats gèniques presenten un baix contingut d'elements repetitius.
Per tal de validar els diferents gens predits pels programes comparem la seqüència predita contra la base de dades d'ESTs emprant el MEGABLAST (donat que estem introduïnt una seqüència llarga i només estem interessades en les correspondències gairebé idèntiques).
Els ESTs (Expressed Sequence Tag) són trossos de seqüència de DNA que oscil.len entre els 200 i els 500 nuclèotids. Aquests fragments s'obtenen a partir de la seqüenciació dels cDNA de diferents gens, d'aquesta manera, acabem obtenint el 5'ESTs i els 3'ESTs. Tots els ESTs obtinguts s'engloben en una bases de dades que conté prop de 3 milions de seqüències que suposen una representació dels gens expressats en determinats teixits de diferents organismes. Els ESTs permetran la validació dels diferents gens predits a partir dels programes a partir de la similaritat entre bases.
Per tal de fer el BLAST per validar els gens predits per cadascún dels programes, cal dividir la seqüència emmascarada (hosmasked.fa) en diferents regions. Així doncs, fem diversos fragments en funció dels gens predits en aquella regió deixant 1000nt de marge 5' i 3' del tros escollit.
- Editem un fitxer que anomenem myseqs.tbl el qual conté els diversos fragments (inici i final) a partir del qual el fastachunk escindirà les diferents regions.
- Aleshores cridem la següent comanda:
egrep -v '^\#' myseqs.tbl | while read REG INI END; do { LEN=`expr $END - $INI + 1`; echo "#--> "$REG"--->"$INI"<-->"$LEN"<--";
( echo ">"$REG"."$INI"-"$LEN ; fastachunk hoxmasked.fa $INI $LEN | fold -60 ) > blast.$REG.fa; }; done
| Regió | Coordenades | Gens predits |
1
(blast.reg1.fa) |
3260-6011 |
gen 1 GENEid
gen 1 GENSCAN
gen 1 FGENESH
|
2
(blast.reg2.fa) |
64008-66430 |
gen 2 GENEid
gen 2 FGENESH
|
3
(blast.reg3.fa) |
104837-171728 |
gen 3, 4 i 5 GENEid
gen 3 i 4 GENSCAN
gen 3 FGENESH
|
4
(blast.reg4.fa) |
175539-218751 |
gen 6, 7, 8 i 9 GENEid
gen 5, 6 i 7 GENSCAN
gen 4, 5, 6, 7 i 8 FGENESH
|
5
(blast.reg5.fa) |
222934-324701 |
gen 10, 11, 12, 13, 14, 15, 16 i 17 GENEid
gen 8, 9, 10 i 11 GENSCAN
gen 9, 10, 11, 12, 13, 14 i 15 FGENESH
|
6
(blast.reg6.fa) |
324130-333644 |
gen 18 GENEid
gen 12 GENSCAN
gen 17 FGENESH
|
7
(blast.reg7.fa) |
350431-425150 |
gen 19 GENEid
gen 13 GENSCAN
gen 17 FGENESH
|
8
(blast.reg8.fa) |
461161-497616 |
gen 14 GENSCAN
gen 18 FGENESH
|
Taula 2. Regions seleccionades
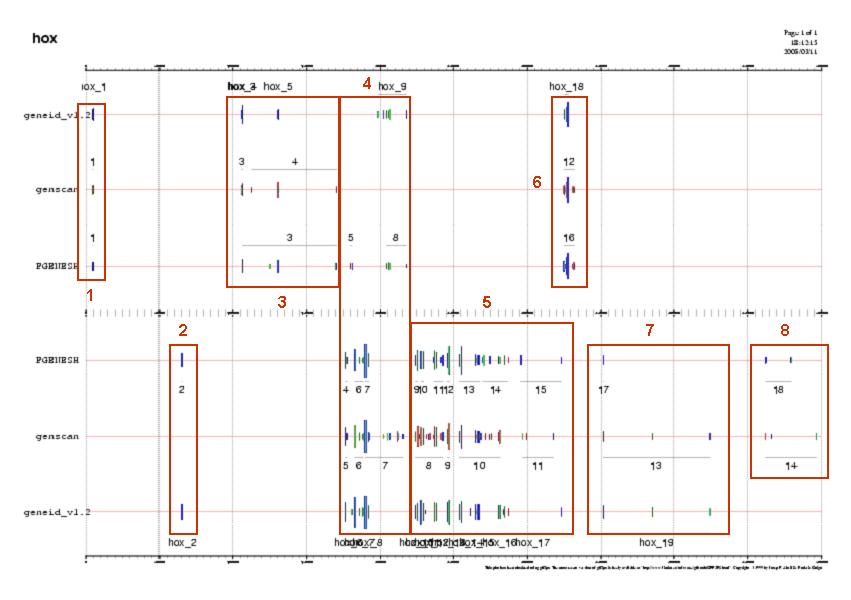
Figura 12. Regions seleccionades
Ja tenim la regió preparada per tal de córrer el megaBLAST usant l'NCBI BLAST server.
Procedim a realitzar el megaBLAST amb els següents paràmetres:
- Base de dades: Est_human
- Organisme: Homo sapiens
- Aligment view: Pairwise
Deixem la resta d'opcions del BLAST amb la configuració per defecte.
- Obtenim els fitxers en format .html i .est:
blast.reg1.html
blast.reg2.html
blast.reg3.html
blast.reg4.html
blast.reg5.html
blast.reg6.html
blast.reg7.html
blast.reg8.html
blast.reg1.est
blast.reg2.est
blast.reg3.est
blast.reg4.est
blast.reg5.est
blast.reg6.est
blast.reg7.est
blast.reg8.est
Per tal d'analitzar els resultats del BLAST cal obtenir un fitxer final d'imatge (.png). Per aconseguir aquest, cal passar l'output del BLAST (format .est) a .gff (mitjançant el parseblast), posteriorment el passarem imatge (amb el gff2ps i la seva conversió a .png).
- Transformem els outputs del BLAST (.est) a .gff mitjançant el programa Parseblast:
parseblast.pl -G blast.reg*.est > blast.reg*.gff
Obtenim els següents resultats:
blast.reg1.gff
blast.reg2.gff
blast.reg3.gff
blast.reg4.gff
blast.reg5.gff
blast.reg6.gff
blast.reg7.gff
blast.reg8.gff
- A continuació cal corregir les coordenades, és a dir, cal convertir les coordenades relatives a absolutes a la seqüència, dels fitxers .gff.
gawk 'BEGIN{OFS="\t"}{$4=$4+3260; $5=$5+3260;print}' blast.reg1.gff > blast.reg1.gff.absolut
gawk 'BEGIN{OFS="\t"}{$4=$4+64008; $5=$5+64008;print}' blast.reg2.gff > blast.reg2.gff.absolut
gawk 'BEGIN{OFS="\t"}{$4=$4+104837; $5=$5+104837;print}' blast.reg3.gff > blast.reg3.gff.absolut
gawk 'BEGIN{OFS="\t"}{$4=$4+175539; $5=$5+175539;print}' blast.reg4.gff > blast.reg4.gff.absolut
gawk 'BEGIN{OFS="\t"}{$4=$4+222934; $5=$5+222934;print}' blast.reg5.gff > blast.reg5.gff.absolut
gawk 'BEGIN{OFS="\t"}{$4=$4+324130; $5=$5+324130;print}' blast.reg6.gff > blast.reg6.gff.absolut
gawk 'BEGIN{OFS="\t"}{$4=$4+350431; $5=$5+350431;print}' blast.reg7.gff > blast.reg7.gff.absolut
gawk 'BEGIN{OFS="\t"}{$4=$4+461161; $5=$5+461161;print}' blast.reg8.gff > blast.reg8.gff.absolut
- El pas posterior per a la visualització dels diferents fitxers consisteix a passar els outputs .gff a .ps (gff2ps) i aquests a .png, finalment visualitzem els fitxers obtinguts amb el Kview.
blast.reg1.png
blast.reg2.png
blast.reg3.png
blast.reg4.png
blast.reg5.png
blast.reg6.png
blast.reg7.png
blast.reg8.png
NOTA: Només hem posat el link d'aquelles regions que amb els paràmetres inicials de cerca obteniem spliced ESTs.
Gràcies a la següent comanda podrem comptar quants ESTs singulars trobem per a cada predicció. La presència d'aquests ESTs singulars no ens donen informació per a la validació donat que poden ser artefactes produïts durant la construcció de les llibreries d'ESTs, així doncs, només els spliced ESTs són considerats com a evidència de la presència de gens.
cut -f 9 blast.reg*.gff.absolut | sort | uniq -c | sort -r -k 1,1 | more
Observem que amb els paràmetres introduïts inicialment en el BLAST obtenim moltes regions amb ESTs singulars i, per tant, cal procedir a repetir el BLAST donat que ens interessen els spliced ESTs com hem citat anteriorment.
Regions en les quals obtenim únicament ESTs singulars
blast.reg1.gff.absolut
blast.reg3.gff.absolut
blast.reg7.gff.absolut
blast.reg8.gff.absolut
Regions en les que tenim spliced ESTs
blast.reg2.gff.absolut
blast.reg4.gff.absolut
blast.reg5.gff.absolut
blast.reg6.gff.absolut
Observem que la meitat de les regions contenen ESTs singulars. Dels resultats obtinguts deduïm que probablement el que succeeix en aquest cas és que les llibreries d'ESTs són incompletes, donat que estem analitzant la regió que conté el cluster dels gens hox, que com hem citat en la introducció són gens que s'expressen en moments determinats del desenvolupament i, per tant, és difícil aconseguir-ne una llibreria completa.
A continuació procedim a realitzar el megaBLAST relaxant els paràmetres de cerca per tal d'aconseguir spliced ESTs. Primer començarem per reduir la mida de la paraula, continuarem rebaixant la identitat fins a un 95% i si amb aquests paràmetres encara no trobem spliced ESTs farem la cerca en primats, mamífers i en última instància en vertebrats. A més a més, també augmentem el número de descriptors fins a 1000 i els alinineaments fins a 250 per tal d'obtenir la màxima informació.
Seleccionant les opcions:
- Mida de la paraula: 11.
- Percentatge d'identitat: 95%.
En les regions 1 (blast2.reg1.html), 7 (blast2.reg7.html) i 8 (blast2.reg8.html) no trobem evidència d'spliced ESTs.
A la regió 3 augmentant el número de descriptors a 1000 i els aliniaments a 250 obtenim spliced ESTs (blast2.reg3.html), blast2.reg3.est) . Cal repetir el procediment anterior per tal d'obtenir el fitxer definitiu de la regió 3 en format .gff (blast2.reg3.gff) i les coordenades corregides (blast2.reg3.gff.absolut)
Així doncs, concloïm que les regions 1, 7 i 8 no podran ser validades per ESTs.
Finalment per a considerar únicament els spliced ESTs, hem d'identificar aquells hsp que apareguin més d'una vegada. Partint dels fitxers *.gff.absolut executem la següent comanda per tal d'eliminar els ESTs singulars.
gawk -f getsplicedhsp.awk blast.reg2.gff.absolut > blast.reg2.spliced.gff
gawk -f getsplicedhsp.awk blast2.reg3.gff.absolut > blast2.reg3.spliced.gff
gawk -f getsplicedhsp.awk blast.reg4.gff.absolut > blast.reg4.spliced.gff
gawk -f getsplicedhsp.awk blast.reg5.gff.absolut > blast.reg5.spliced.gff
gawk -f getsplicedhsp.awk blast.reg6.gff.absolut > blast.reg6.spliced.gff
En aquest punt podem visualitzar les prediccions gèniques juntament amb els ESTs.
- El primer pas consisteix en eliminar el marc de lectura dels ESTs per tal de permetre una correcta visualització, és a dir, a l'eliminar el frame farem que els ESTs es mostrin a la part central del dibuix, i els gens predits en forward a la regió superior i en revers en la inferior.
gawk '{$7 = "."; print $0}' blast.reg2.spliced.gff > blast.reg2.spliced.noframe.gff
gawk '{$7 = "."; print $0}' blast2.reg3.spliced.gff > blast2.reg3.spliced.noframe.gff
gawk '{$7 = "."; print $0}' blast.reg4.spliced.gff > blast.reg4.spliced.noframe.gff
gawk '{$7 = "."; print $0}' blast.reg5.spliced.gff > blast.reg5.spliced.noframe.gff
gawk '{$7 = "."; print $0}' blast.reg6.spliced.gff > blast.reg6.spliced.noframe.gff
- Procedim a convertir els fitxers .gff a .ps amb el gff2ps, arrodonint els límits de les regions a múltiples de 100.
gff2ps -S 64000 -E 66500 hoxmasked.geneid.gff hoxmaskedp.genscan.gff hoxmasked.fgenesh.gff blast.reg2.spliced.noframe.gff > regio2estgens.ps
gff2ps -S 104800 -E 171800 hoxmasked.geneid.gff hoxmaskedp.genscan.gff hoxmasked.fgenesh.gff blast2.reg3.spliced.noframe.gff > regio3estgens.ps
gff2ps -S 175500 -E 218800 hoxmasked.geneid.gff hoxmaskedp.genscan.gff hoxmasked.fgenesh.gff blast.reg4.spliced.noframe.gff > regio4estgens.ps
gff2ps -S 222900 -E 324700 hoxmasked.geneid.gff hoxmaskedp.genscan.gff hoxmasked.fgenesh.gff blast.reg5.spliced.noframe.gff > regio5estgens.ps
gff2ps -S 324100 -E 333700 hoxmasked.geneid.gff hoxmaskedp.genscan.gff hoxmasked.fgenesh.gff blast.reg6.spliced.noframe.gff > regio6estgens.ps
- Convertim el post script a format .png per tal de visualitzar-ho amb el Kview.
convert -density 150 -rotate 90 regio2estgens.ps regio2estgens.png
convert -density 150 -rotate 90 regio2estgens.ps regio3estgens.png
convert -density 150 -rotate 90 regio2estgens.ps regio4estgens.png
convert -density 150 -rotate 90 regio2estgens.ps regio5estgens.png
convert -density 150 -rotate 90 regio2estgens.ps regio6estgens.png
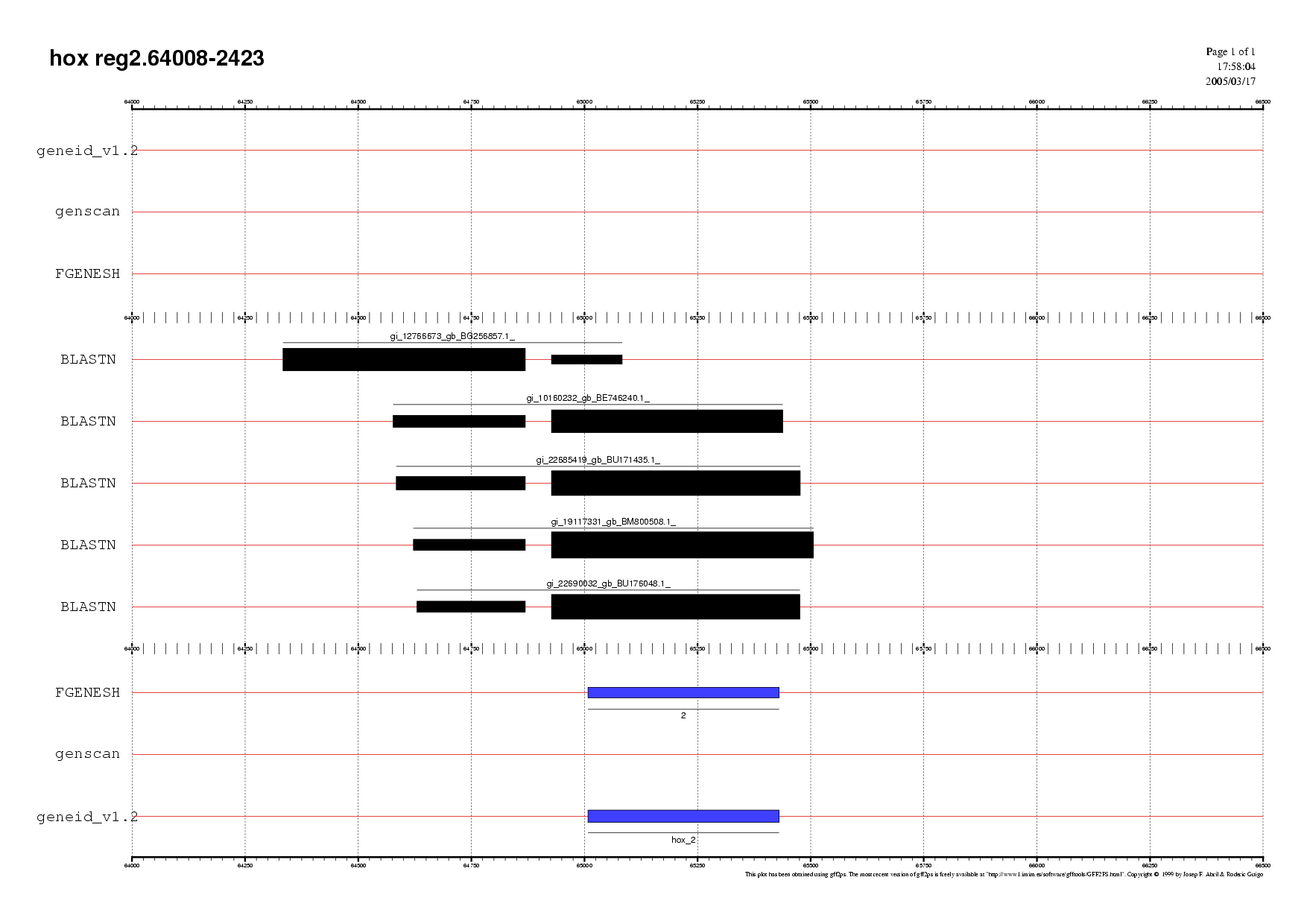
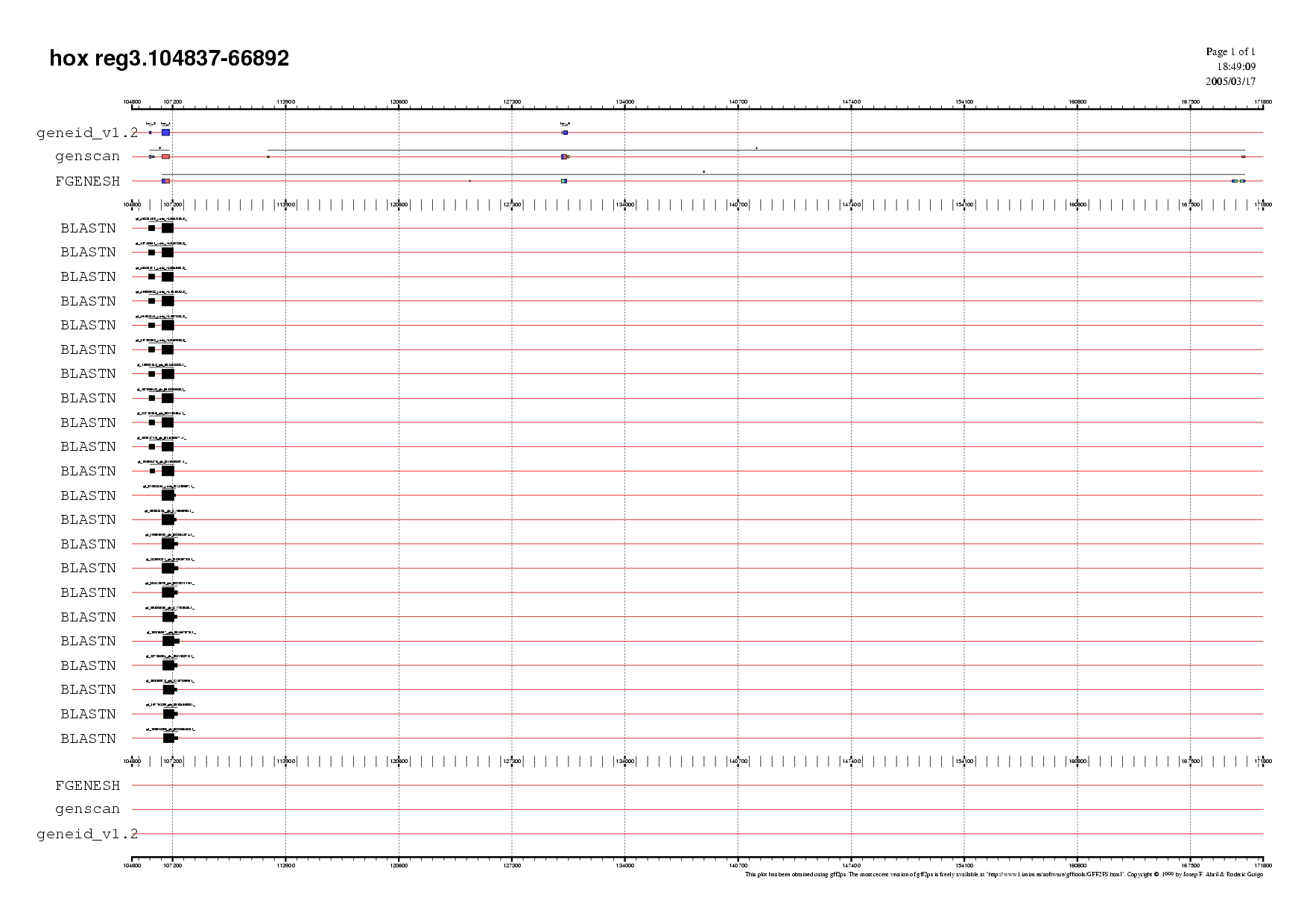
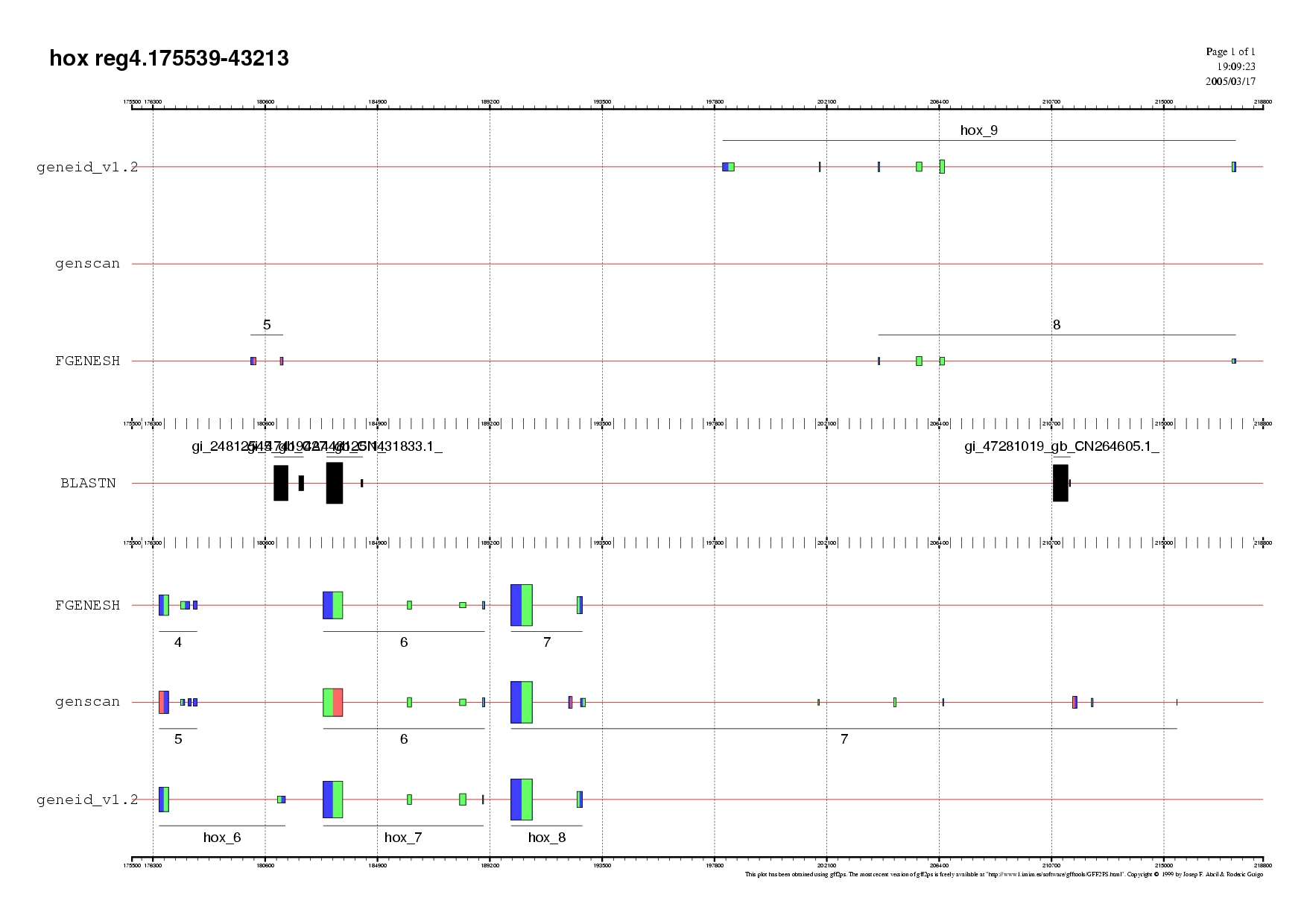
Figura 13. Validació per ESTs de la regió 2 Figura 14. Validació per ESTs de la regió 3 Figura 15. Validació per ESTs de la regió 4
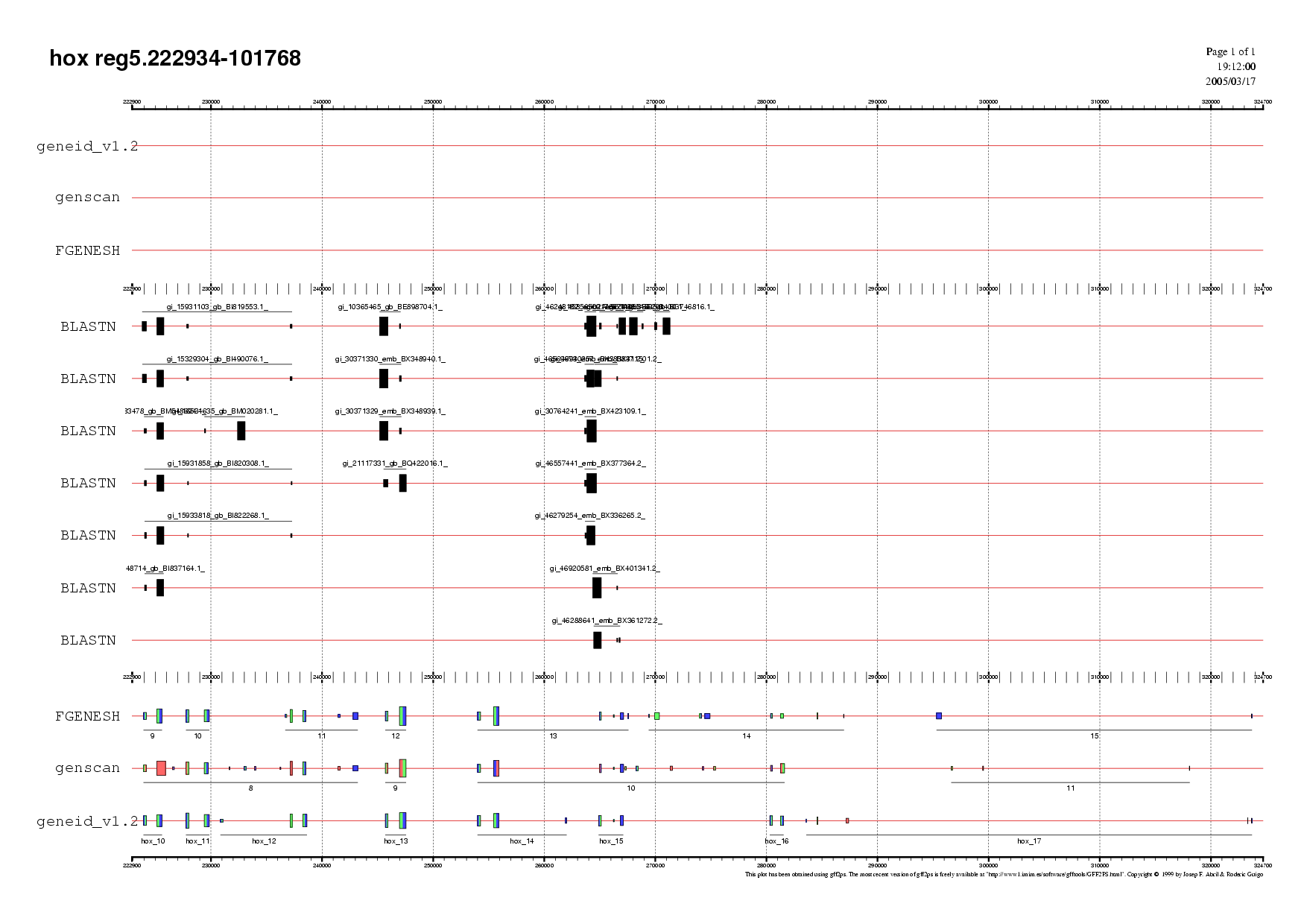
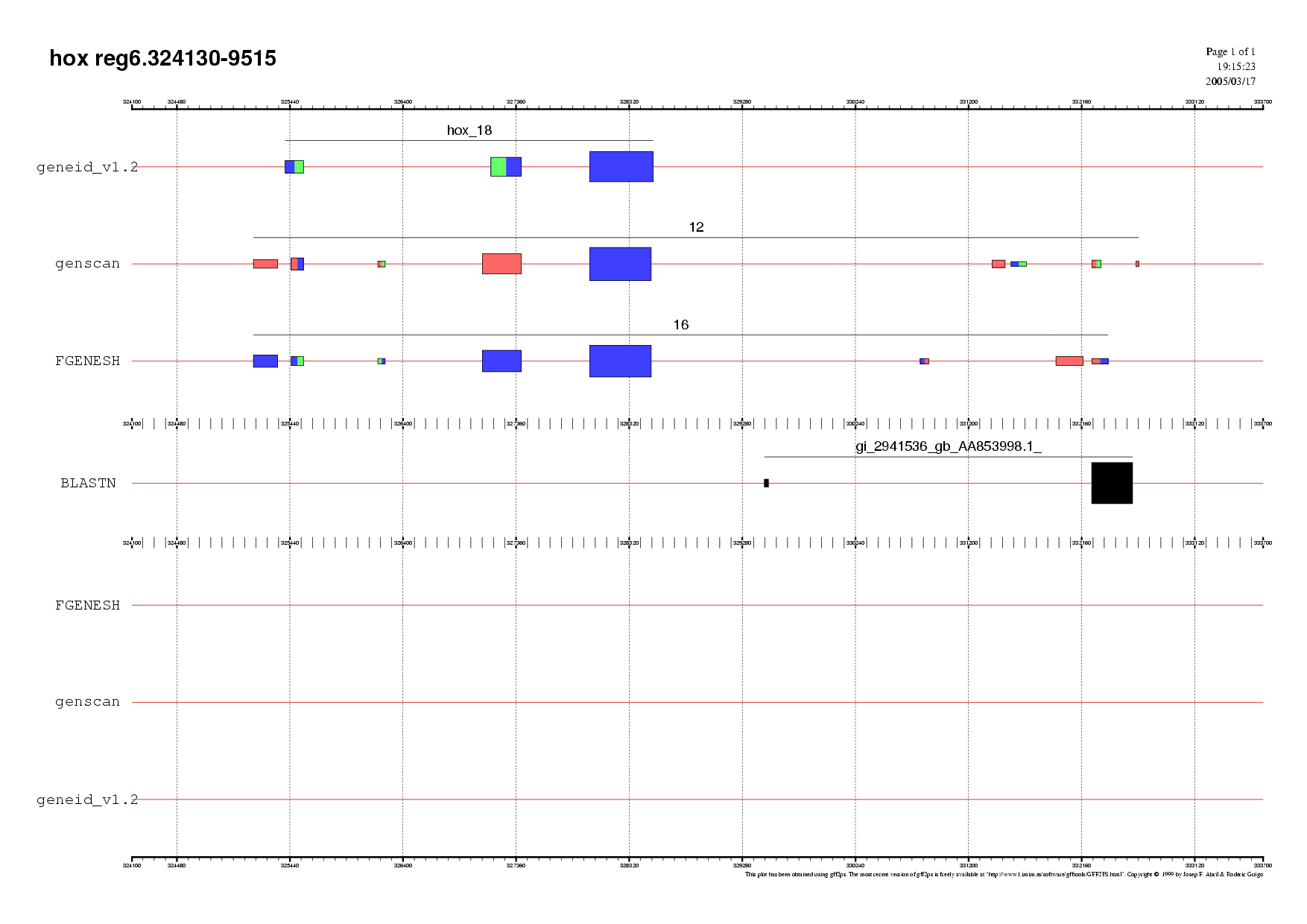
Figura 16. Validació per ESTs de la regió 5 Figura 17. Validació per ESTs de la regió 6
Selecció de gens
Els gens seleccionats els podem englobar bàsicament en tres categories:
1- Gens predits per més d'un programa i validats per ESTs.
2- Gens predits per més d'un programa la validació dels quals no està suportada per ESTs.
3- Gens predits exclusivament per un programa.
Globalment podem considerar que la seqüència analitzada no presenta un bon suport per ESTs, en molts casos els ESTs no suporten tots els exons predits i, fins i tot, hi ha regions en les quals no n'hem trobat cap evidència.
A) Regió 1
En aquesta regió observem un gen (reverse) que està predit pels tres programes (gen 1 GENEid, GENSCAN, FGENESH), però cap dels seus exons està suportat per ESTs, aquest fet però no suposa que la predicció de gens "ab initio" sigui incorrecta, és a dir, pot succeir que aquests gens s'expressin en condicions restringides i que, per tant, no estiguin dipositats en llibreries d'ESTs.
Per tant, caracteritzarem la proteïna (proteïna 1) codificada per aquest gen.
B) Regió 2
Aquesta regió engloba els gen 2 predit per GENEid i per FGENESH (reverse). Es tractaria d'un gen que segons els programes de predicció de gens només tindria un exó, però al fer la validació per ESTs n'observem dos, és a dir, podria ser que els programes només haguéssin predit un exó del gen. Aquest gen codifica per la proteïna 2.
C) Regió 3
En aquesta regió tots els gens estan en forward.
- Gen 3 i 4 del GENEid - Gen 3 GENSCAN: El GENSCAN considera els gens 3 i 4 del GENEid com un sol gen amb dos exons. Quan fem la validació per ESTs, optem pel gen 3 del GENSCAN donat que els ESTs suporten els dos exons d'un mateix gen. La proteïna predita és la proteïna 3.
- Gen 4 del GENSCAN: Desestimem la seva predicció donat que no està suportat per ESTs.
- Gen 5 del GENEid: Desestimem la seva predicció donat que no està suportat per ESTs.
- Gen 3 del FGENESH: tot i que només un exó està suportat per ESTs el seleccionem per tal de realitzar els anàlisis posteriors. La proteïna predita l'anomenem proteïna 4.
D) Regió 4:
- Gens en Forward:
- Gen 9 GENEid - Gen 8 FGENESH: En aquests gens no hi tenim evidència d'ESTs però comparteixen diversos exons. Decidim quedar-nos amb el gen 9 del GENEid, per què aquest gen engloba tots els exons predits pel FGENESH. La proteïna resultant l'anomenem proteïna 5.
- Gen 5 FGENESH: Descartem aquesta predicció donat que només està predit per un programa i no està suportat per ESTs.
- Gens en reverse:
- Gen 5 GENSCAN - Gen 4 FGENESH - Gen 6 GENEid: Aquestes prediccions comparteixen el mateix exó inicial, el gen 4 FGENESH i el gen 5 GENSCAN, a més a més, comparteixen d'altres exons. Així doncs, de les tres prediccions escollim el gen 4 d'FGENESH (marcs de lectura correctes). Aquest gen codifica per la proteïna que anomenem proteïna 6.
- Gen 6 GENSCAN - Gen 7 GENEid - Gen 6 FGENESH: Aquests gens han estat igualment predits per tots els programes. Ens quedem amb el gen 7 del GENEid (el gen 6 del GENSCAN observem que no té el marc de lectura correcte i, per tant, l'obviem). Aquest gen codifica per la proteïna 7.
- Gen 7 GENSCAN - Gen 7 FGENESH - Gen 8 GENEid: el gen 7 de FGENESH és igual al 8 del GENEid, és un gen compost per dos exons, en canvi el GENSCAN ha predit un gen més llarg que comparteix els dos primers exons amb la resta de prediccions. Però com que la predicció majoritària ha estat la d'un gen amb dos exons, ens quedem amb 7 FGENESH o 8 GENEid. Aquest gen codifica per la proteïna 8.
E) Regió 5:
En aquesta regió tots els gens són en reverse.
- Gen 9 FGENESH i gen 10 GENEid: es tracta del mateix gen i es troba validat per ESTs, per tant, el seleccionem (proteïna 9).
- Gen 10 FGENESH i 11 GENEid: Es tracta del mateix gen predit per dos programes i, a més a més, està validat per ESTs. El gen codifica per la proteïna 10.
- Gen 8 GENSCAN: aquest gen engloba 3 gens que prediu l'FGENESH i 3 més predits pel GENEid. A més a més, aquest gen es troba validat per ESTs. Així doncs, podem afirmar que la validació és força consistent, veiem que la predicció és suportada per 3'ESTs, tot i que no està suportada per 5'ESTs, però com que degut al mecanisme pel qual s'obtenen els ESTs, els 3'ESTs són més fiables que els 5'ESTs (si trobem només 5'ESTs no podem estar segurs de l'extrem 5' del gen donat que es pot extendre més enllà). El gen seleccionat codifica per la proteïna 11).
- Gen 9 GENSCAN, 13 GENEid, 12 FGENESH: Es tracta d'un gen predit (codifica per la proteïna 12) pels tres programes i, a més a més, aquesta predicció està suportada per ESTs.
- Gen 13 FGENESH: Analitzem aquest gen tot i que està validat per 5'ESTs. Codifica per la proteïna 13.
- Gen 14 FGENESH: Seleccionem aquest gen donat que té un exó que tot i estar validat per ESTs, no s'ha predit per cap altre programa més. La proteïna per la qual codifica és la proteïna 14.
- Gen 14, 15, 16 GENEid: no els incloem en l'anàlisi (aquests gens són diferents exons s'inclouen en altres prediccions).
- Gen 15 FGENESH, 11 GENSCAN, 17 GENEid: no els incloem a la cerca donat que no estan validats per ESTs i a més a més només són predits per un programa.
F) Regió 6:
Aquesta regió engloba 3 gens en forward.
- Gen 18 GENEid: l'omitim de la selecció que només el prediu aquest programa i a més a més no està suportat per ESTs. A més a més, els exons estàn inclosos en d'altres prediccions.
- Gen 12 GENSCAN - 16 FGENESH: Ens quedem amb el gen 16 d'FGENESH, donat que prediu exons similars al 12 de GENSCAN i l'FGENESH és el que té el marc de lectura correcte. Aquest gen codifica per la proteïna 15.
G) Regió 7:
En aquesta regió no hem trobat evidència d'spliced ESTs i, per tant, per a la selecció de gens (tots ells en reverse) ens basarem en la comparació de prediccions pels diferents programes.
- Gen 17 FGENESH: no l'incloem en l'anàlisi donat que no està recolzat per cap altre programa.
- Gen 13 GENSCAN i Gen 19 GENEid: ambdós programes prediuen gens similars, per això, els incloem en la selecció. El gen 13 de GENSCAN codifica per la proteïna 16.
En un inici havíem seleccionat per l'anàlisi el gen 19 predit per GENEid (codifica per la proteïna, al observar que es tracta d'una predicció parcial, la proteïna predita no s'inicia en un un exó inicial sinó en un d'intern (no comença per metionina). Per això, es decideix finalment no incloure-la en l'anàlisi.
E) Regió 8:
En aquesta regió, de la mateixa manera que en el cas anterior, no s'ha trobat evidència d'ESTs. Per tant, compararem els diversos gens que prediuen els diferents programes.
En aquesta regió trobem el gen 18 d'FGENESH i el gen 14 de GENSCAN. Ambdós gens predits només comparteixen un exó, però, els seleccionem, donat que en aquesta regió només s'han predit aquests dos gens.
- El cas del gen 14 del GENSCAN es tracta d'una predicció parcial, la proteïna no s'inicia per Metionina i, per tant, es decideix no incloure'l finalment en l'anàlisi.
En aquest apartat procedim a caracteritzar les proteïnes predites, mitjançant el BLASTP, el que es fa es comparar la seqüència aminoacídica obtinguda contra un conjunt de proteïnes conegudes.
Així doncs, emprarem la base de dades Swissprot i seleccionem "Humà" com a organisme, si no hi trobem aliniaments, procedim a realitzar la cerca en d'altres organismes (primats i, posteriorment, rossegadors).
| Proteïna | Gen/s corresponent |
| 1 |
gen 1 GENEid
gen 1 GENSCAN
gen 1 FGENESH |
| 2 |
gen 2 FGENESH
gen 2 GENEid
|
| 3 |
gen 3 GENSCAN |
| 4 |
gen 3FGENESH |
| 5 |
gen 9 GENEid |
| 6 |
gen 4 FGENESH |
| 7 |
gen 7 GENEid |
| 8 |
gen 7 FGENESH
gen 8 GENEid |
| 9 |
gen 9 FGENESH
gen 10 GENEid |
| 10 |
gen 10 FGENESH
gen 11 GENEid |
| 11 |
gen 8 GENSCAN |
| 12 |
gen 12 FGENESH
gen 9 GENSCAN
gen 13 GENEid |
| 13 |
gen 13 FGENESH |
| 14 |
gen 14 FGENESH |
| 15 |
gen 16 FGENESH |
| 16 |
gen 13 GENSCAN |
| 17 |
gen 18 FGENESH |
Taula 3. Correlació proteïna-gens predits
A) Proteïna 1:
Score = 237 bits (604), Expect = 1e-62
Identities = 140/249 (56%), Positives = 145/249 (58%), Gaps = 54/249 (21%)
Un gran número de nucleòtids de la proteïna predita s'alineen (un 56%) amb la proteïna ribosomal humana L7a (score:237, E-value: 1e-62).Observem que es tracta d'una proteïna del cromosoma 9, quan a priori esperaríem que s'hagués aliniat amb una proteïna del cromosoma 7, això pot ser degut a que la proteïna predita no hagi estat sequüenciada però que presenti dominis homòlegs a aquesta proteïna del cromosoma 9.
També cal tenir en compte que es tracta d'una proteïna en forward mentre que els diferents programes la predeien en reverse.
Figura 18. Orientació del gen
2) Funció:
Aquest gen codifica per una proteïna ribosomal que forma part del la subunitat 60S del ribosoma. Pertany a la família L7AE de proteïnes ribosomals. A part, pot interaccionar amb diverses subclasses de receptors hormonals i inhibir la seva capacitat de transactivar (inhibeixen la seva unió als elements de resposta del DNA). Com és típic dels gens que codifiquen per proteïnes ribosomals, hi ha múltiples pseudogens d'aquest gen dispersats pel genoma.
3) Dominis conservats:
Observem que aquesta proteïna conté el següent domini conservat.

Figura 19. Domini de la proteïna 1
B) Proteïna 2:
Score = 223 bits (569), Expect = 1e-59
Identities = 109/139 (78%), Positives = 117/139 (84%)
La proteïna predita s'alinia amb la proteïna "High mobility group (HMG-4)" i presenta un score i identitat elevada (78%). A part, podem dir que l'aliniament és significatiu ja que presenta un E-value baix.
La proteïna aliniada però es localitza en el cromosoma X (Xq28), pot succeir com en el cas anterior, que s'alinii perque les proteïines presentin dominis similars.
La nostra proteïna té sentit reverse i aquesta proteïna per contra és en forward.
Figura 20. Orientació del gen
2) Funció:
Les proteïnes HMG són components abundants del nucli dels mamífers i s'engloben en tres famílies. En ratolins s'ha vist que el gen HMG4 és altament expressat en l'embrió, aquests transcrits són gairebé indetectats en els teixits adults, es dedueix que aquesta proteïna és requerida pel desenvolupament. El gen humà HMG4, és extremadament similar al seu homòleg en ratolí, i s'ha seqüenciat en la regió Xq28.
3) Dominis conservats:

Figura 21. Domini de la proteïna 2
C) Proteïna 3:
Score = 200 bits (509), Expect = 2e-52
Identities = 123/246 (50%), Positives = 145/246 (58%), Gaps = 42/246 (17%)
La proteïna s'alinia amb gran part (identitat del 50%) de la cadena 4 alfa de la Tropomiosina (TPM4) i presenta un score superior a 200 bits.
Aquesta proteïna es localitza en el cromosoma 19 (19p13.1) de manera que pot succeir un fet equivalent als dos casos anteriors.
En aquest cas, la proteïna amb la qual s'alinia es troba en forward, la mateixa direcció que la nostra proteïna.
Figura 22. Orientació del gen
2) Funció:
Es tracta d'una isoforma de la tropomiosina la qual és necessària per a la funció contràctil del cor en l'embrió. Aquesta, interacciona selectivament amb formes tant monomèriques com multimèriques de l'actina, incloent els filaments d'actina.
3) Dominis conservats:

Figura 23. Domini de la proteïna 3
D) Proteïna 4:
Score = 184 bits (468), Expect = 4e-47
Identities = 101/144 (70%), Positives = 110/144 (76%)
L'aliniament contra la base de dades de Swissprot seleccionant Homo sapiens no ens dona resultats significatius i, per tant, procedim a seleccionar Primats, però tampoc obtenim bons aliniaments. Procedim a seleccionar vertebrats i tampoc obtenim bons aliniaments.
Al no obtenir aliniaments amb score elevat (>=200) ens fixem en els aliniaments obtinguts per humà amb un score entre 80-200. Tot i no quedar-nos amb un aliniament amb score no molt elevat, observem que la identitat és bastant alta (70%).
La proteïna amb la qual s'alinia és la cadena 3 alfa de la Tropomiosina (TPM3).Es tracta d'una proteïna localitzada en el cromosoma 1 (1q21.2) però segurament surten aliniades perquè comparteixen dominis.
Com ja ens ha passat en altres proteïnes, està en reverse i la proteïna predita en forward.
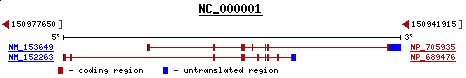
Figura 24. Orientació del gen
3) Dominis conservats:

Figura 25. Domini de la proteïna 4
E) Proteïna 5:
Score = 32.7 bits (73), Expect = 0.19
Identities = 36/171 (21%), Positives = 63/171 (36%), Gaps = 29/171 (16%)
Es realitza el BLASTP seleccionant Homo sapiens, observem com s'alinia una petita part de la nostra proteïna. A part, obtenim un score baix. Si seleccionem rossegadors l'score continua essent baix (similar al de l'aliniament amb proteïnes humanes), per això, decidim quedar-nos amb els aliniaments humans.
Tot i no presentar un score i una identitat òptima i un E value elevat, la nostra proteïna 5 s'alinea amb l'Histona Deacetilasa 4. Aquesta es localitza en el cromosoma 2 (2q37.2) i està en reverse, mentre que la proteïna que s'havia predit està en forward).
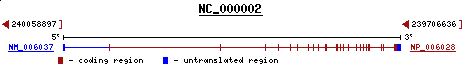
Figura 26. Orientació del gen
2) Funció:
Les histones juguen un paper important en la regulació de la transcripció, el progrés del cicle cel.lular i processos de desenvolupament. L'acetilació i desacetilació de les histones altera l'estructura cromosòmica i afecta l'accés dels factors de transcripció al DNA. L'histona deacetilasa 4 no s'uneix directament al DNA sinó que ho fa a través dels factors de transcripció MEF2C i MEF2D.
3) Dominis conservats:
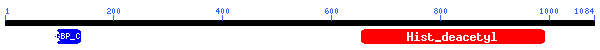
Figura 27. Domini de la proteïna 5
F) Proteïna 6:
Score = 494 bits (1272), Expect = e-140
Identities = 254/330 (76%), Positives = 254/330 (76%), Gaps = 48/330 (14%)
La proteïna predita s'alinia amb bona identitat i score amb l'homeobox protein Hox-A1.
La regió que estem analizant engloba el cluster de gens HOX al cromosoma 7 i la HOX-A1 (proteïna amb la que s'ha aliniat) és una proteïna d'aquest cromosoma (7p15.3). Però, aquesta proteïna és en reverse i el gen de la nostra predicció era en forward.
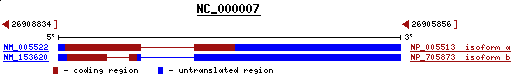
Figura 28. Orientació del gen
2) Funció:
En vertebrats, els gens que codifiquen per la classe de factors de transcripció anomenats gens homeobox es troben en clusters que reben el nom d'A, B, C i D en quatre cromosomes separats. L'expressió d'aquestes proteïnes és regulada espacialment i temporalment durant el desenvolupament embrionari.
La proteïna amb la que s'ha aliniat forma part del cluster A (localitzat al cromosoma 7) i codifica per a un factor de transcripció que regula l'expressió gènica, la morfogènesis i la diferenciació. S'han trobat dues variants de transcripció d'aquest gen codificant dues isoformes diferents, només una d'aquestes isoformes conté la regió d'homeodomini.
3) Dominis conservats:
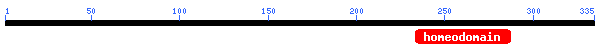
Figura 29. Domini de la proteïna 6
A partir de la base de dades Interpro (dóna informació dels dominis de proteïnes coneguts) hem identificat dominis que ja esperàvem. Podem observar com identifica el domini homeobox (descrit inicialment en Drosophila), el qual és conservat i conegut en molts altres animals (com ratolins,...). A part, ens mostra la similaritat (en seqüència i estructura) amb proteïnes d'unió a DNA (per exemple, proteïnes que presenten el motiu "helix-turn-helix" (HTH).
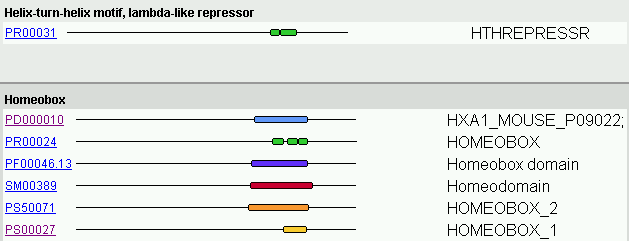
Figura 30. Homeodominis
G) Proteïna 7:
Score = 434 bits (1117), Expect = e-122
Identities = 218/247 (88%), Positives = 218/247 (88%)
La proteïna predita s'alinia amb "Homeobox protein Hox-A2", aquest aliniament té un score i identitat (88%) elevats i un e-value baix, es tracta, doncs, d'un bon aliniament.
La localització d'aquesta proteïna és al cromosoma 7 (7p15-p14), i el gen està orientat en reverse com la nostra predicció.
Figura 31. Orientació del gen
2) Funció
3) Dominis conservats:

Figura 32. Domini de la proteïna 7
Com en la proteïna anterior, anem a la base de dades d'Interpro i obtenim la mateixa informació que la proteïna anterior.
H) Proteïna 8:
Score = 402 bits (1033), Expect = e-113
Identities = 214/316 (67%), Positives = 220/316 (69%), Gaps = 9/316 (2%)
La proteïna predita s'alinia amb "Homeobox protein Hox-A3 (Hox-1E)", els paràmetres d'score, e-value, i identitat ens mostren que l'aliniament és bo.
Aquesta proteïna està localitzada en el cromosoma 7 (7p15-p14) i està en reverse com la nostra proteïna.
Figura 33. Orientació del gen
2) Funció
3) Dominis conservats:

Figura 34. Domini de la proteïna 8
Com hem fet en altres proteïnes, cerquem en la base de dades d'Interpro i obtenim la mateixa informació que abans.
I) Proteïna 9:
Score = 321 bits (822), Expect = 1e-88
Identities = 160/231 (69%), Positives = 160/231 (69%)
La proteïna s'ha aliniat amb "Homeobox protein Hox-A5 (Hox-1C)". A part, està orientada en reverse com la predicció, i es troba en el cromosoma 7 (7p15-p14).

Figura 35. Orientació del gen
2) Funció
3) Dominis conservats:
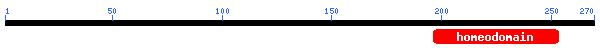
Figura 34. Domini de la proteïna 9
Com en totes les proteïnes que han detectat homeodominis, anem a la base de dades Interpro i observem com troba similitud amb els homeodominis de Drosophila, en concret amb un grup de gens que reben el nom d'Antennapedia. Aquest grup s'encarrega de regular la formació de les estructures de la pota en Drosophila. Aquest grup és important ja que va ser un dels primers gens homeòtics estudiats i va permetre el descobriment del domini homeobox.
Figura 35. Homeodominis
J) Proteïna 10:
Score = 458 bits (1178), Expect = e-130
Identities = 220/233 (94%), Positives = 220/233 (94%)
Aquesta proteïna s'alinia amb "Homeobox protein Hox-A6 (Hox-1B)", l'e-value és baix, l'score és elevat i les identitats són elevades, es tracta d'un aliniament òptim.
Observem que es localitza al cromosoma 7 (7p15-p14) i que el gen té una orientació reverse com la proteïna predita.

Figura 36. Orientació del gen
2) Funció
3) Dominis conservats:
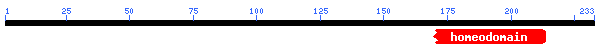
Figura 37. Domini de la proteïna 10
K) Proteïna 11:
Score = 404 bits (1039), Expect = e-113
Identities = 199/270 (73%), Positives = 199/270 (73%)
La proteïna s'alinia amb "Homeobox protein Hox-A5 (Hox-1C)" presentant un score i identitat elevats i e-value baix, per tant, podem considerear que l'aliniament és correcte.
Es localitza al cromosoma 7 (7p15-p14) i té orientació reverse com la proteïna predita.

Figura 38. Orientació del gen
2) Funció
3) Dominis conservats:
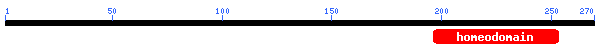
Figura 39. Domini de la proteïna 11
Com hem anat fent amb les proteïnes que presenten homeodominis, cerquem homologia amb la base de dades Interpro i obtenim, com ja ha passat amb la proteïna 9 i 10 similitud amb el complex de gens Antennapedia de Drosophila.
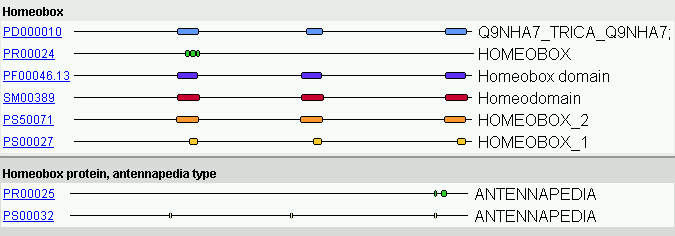
Figura 40. Homeodominis
L) Proteïna 12:
Score = 481 bits (1239), Expect = e-137
Identities = 236/272 (86%), Positives = 236/272 (86%)
Aquesta proteïna s'ha aliniat amb "Homeobox protein Hox-A9 (Hox-1G)", amb un score elevat, e-value baix i identitat elevada, és un aliniament correcte.
Es localitza al cromosoma 7 (7p15-p14) i el seu gen té una orientació reverse com la nostra proteïna.
Figura 41. Orientació del gen
2) Funció
3) Dominis conservats:

Figura 42.Domini de la proteïna 12
Al fer l'Interpro obtenim els mateixos resultats que en el cas anterior.
M) Proteïna 13:
Score = 386 bits (991), Expect = e-108
Identities = 195/250 (78%), Positives = 195/250 (78%)
Aquesta proteïna s'alinea amb "Homeobox protein Hox-A10 (Hox-1H)". L'aliniament té l'score elevat, l'e-value es baix i les identitats són elevades es tracta d'un aliniament òptim.
Es troba en el cromosoma 7 (7p15-p14) en una orientació reverse com la proteïna predita.

Figura 43. Orientació del gen
2) Funció
3) Dominis conservats:

Figura 44.Domini de la proteïna 13
Al fer l'Interpro obtenim els següents resultats:

Figura 45. Homeodominis
N) Proteïna 14:
Score = 320 bits (821), Expect = 6e-88
Identities = 149/149 (100%), Positives = 149/149 (100%)
La proteïna s'ha alineat amb "Homeobox protein Hox-A13 (Hox-1J)" amb score i identitat elevada i e-value inferior, es tracta d'un bon aliniament.
Es troba al cromosoma 7 (7p15-p14)en reverse com la proteïna predita.

Figura 46. Orientació del gen
2) Funció
3) Dominis conservats:
Figura 47.Domini de la proteïna 14
Al fer l'Interpro obtenim els següents resultats:

Figura 48. Homeodominis
O) Proteïna 15:
Score = 597 bits (1538), Expect = e-171
Identities = 315/447 (70%), Positives = 315/447 (70%), Gaps = 83/447 (18%)
La proteïna s'ha aliniat amb "Homeobox even-skipped homolog protein 1 (EVX-1)", aquest aliniament presenta un score elevat i un e-value baix, juntament amb una identitat elevada, es tracta doncs d'un bon aliniament.
Es troba al cromosoma 7 (7p15-p14) i es troba en forward com la proteïna predita.
Figura 49. Orientació del gen
2) Funció:
Aquest gen codifica per una proteïna que és membre de la família homeo box relacionat a Eve de vertebrats. Aquesta proteïna actua com un repressor transcripcional. Una proteïna similar en ratolí és crítica per a l'embriògenesi temprana.
3) Dominis conservats:

Figura 50. Domini de la proteïna 15
Al fer l'Interpro obtenim els següents resultats:

Figura 51. Homeodominis
O) Proteïna 16:
Score = 70.5 bits (171), Expect = 5e-13
Identities = 36/44 (81%), Positives = 38/44 (86%)
La proteïna s'alinea amb "60S ribosomal protein L35", l'score però és baix, per tant, no es tracta d'un aliniament òptim.
Es tracta d'una gen del cromosoma 9 (9q34.1), en canvi, s'està analitzant una regió del cromosoma 7, pot ser degut a que la proteïna predita contingui dominis equivalents a aquesta proteïna, es tracta d'un gen en reverse com la nostra proteïna.
Figura 51. Orientació del gen
2) Funció:
Aquest gen codifica per una prote&iulm;na que és membre de la família homeo box relacionat a eve de vertebrats. Aquesta prote&iulm;na actua com un repressor transcripcional. Una proteïna similar en ratolí és critica per a l'embrigènesi temprana.
O) Proteïna 17:
Score = 33.5 bits (75), Expect = 0.014
Identities = 16/62 (25%), Positives = 31/62 (50%)
La proteïna s'alinea amb "Matrix metalloproteinase-21 precursor (MMP21)", l'score i la identitat són molt baixos, pel que deduim que no es tracta d'un bon aliniament.
Es tracta d'una gen del cromosoma 10 (10q26.2), l'aliniament pot ser degut a que la proteïna predita contingui dominis equivalents a aquesta proteïna, es tracta d'un gen en reverse com la nostra proteïna.
Figura 52. Orientació del gen
2) Funció:
Aquest gen codifica per un membre de la família de les "matrix metalloproteinase", estan involucrades en el trencament de la matriu extracel.lular en processos fisiològics (desenvolupament embrionari, reproducció...) i patològics (asma i metàstasi).
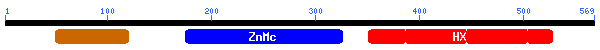
Figura 53. Domini de la proteïna 17
Realització d'una base de dades de proteïnes
En aquest punt es realitza una base de dades de les proteïnes predites per tal d'observar si aquestes formen un cluster.
Com ja s'ha dit els gens Homeobox codifiquen per factors de transcripció els quals es disposen en clusters, anomenats A, B, C i D. La nostra seqüència engloba el cluster HOXA. Aquests gens contenen una seqüència homeobox, és un domini conservat de DNA d'uns 180 parells de bases. L'homeobox codifica per l'homeodomini (d'uns 60 aa) que és una regió responsable de la unió al DNA. Cal dir també que alguns gens homeobox, no són gens homeòtics, l'homeobox és una seqüència motiu, mentre que el terme "homeòtic" es refereix a una descripció funcional dels gens causants de transformacions homeòtiques.
Així doncs, el que es fa es realitzar aliniaments entre totes les proteïnes predites contra elles mateixes, com que els gens Hox contenen l'homeodomini conservat aquests s'aliniaran i es farà evident la presència d'un cluster gènic.
El procediment que seguim és el següent:
- Cal construir un fitxer que contingui les proteïnes que formaran part de la base de dades.
- Un cop tenim el fitxer qeu conté el conjunt de proteïnes procedim a fer el formatdb.
formatdb -p T -i proteines.fa
blastall -p blastp -d proteines.fa -i proteines.fa -e 0.1 > protblast_all.out
parseblast.pl -S -G -i protblast_all.out | sort > protblast_all.gff
- Obtenim un fitxer en format gff, que ens indica com s'han aliniat entre si les proteïnes. Evidentment, ens interessa eliminar aquells aliniaments d'una proteïna amb ella mateixa. Ho aconseguim de la següent manera:
sed 's/;//g;' protblast_all.gff | gawk '$1 != $9' > protblast_all_filtrat.gff
Un cop obtenim el fitxer, procedim a analitzar les dades i observem com una gran part de proteïnes s'alineen entre elles en una o més regions. D'aquest aliniament, resaltem l'aliniament que es produeix entre la proteïna 3 i la 4, resultat lògic donat que cadascuna de les proteïnes codifica per diferents isoformes de la tropomiosina (TPM).
Però sobretot, cal destacar l'aliniament que es produeix entre la proteïna 11 i gran part de la resta de proteïnes de la base de dades (en concret, la proteïna 6, 7, 8, 9, 10, 12, 13, 14, 15). Aquest grup de proteïnes coincideixen amb les que hem trobat que presenten homedominis, el que ens fa pensar que só les proteïnes que formen part del cluster HOXA. També cal resaltar l'aliniament entre la proteïna 11 i la 13, que s'alineen en sis punts diferents.
Al finalitzar l'anàlisi de la regió del cluster HOX A del genoma podem extreure les següents conclusions:
- En emmascarar la seqüència observem que 174803 pb han estat emmascarades, aquestes bases corresponen a regions repetitves (LINES, SINES...).
- En fer un gràfic on s'hi representen conjuntament les prediccions de gens pels tres programes i les regions repetitives observem que les regions amb alta densitat de gens, no contenen elements repetitius.
- Els diferents programes (GENSCAN, GENEid, FGENESH) han predit diferent número de gens. El GENEid és el programa que n'ha predit més (19), l'FGENESH (18) i el GENSCAN n'és el que n'ha predit menys(13).
És important remarcar que dos gens han estat predits exactament pels tres programes: el gen 1 GENEid, GENSCAN i FGENESH i el 13 de GENEid, el 9 de GENSCAN i el 12 d'FGENESH.
També cal dir que si bé, només aquests dos gens tenen correspondència exacta entre els 3 programes, hi ha d'altres gens en que si bé la correspondència no és total, les coordenades inicials coincideixen en diferents programes. És a dir, hi ha programes que consideren exons d'un mateix gen, en canvi d'altres que ho consideren com a diferents gens.
- Posteriorment es compara la seqüència predita contra la base de dades d'ESTs per tal de validar les prediccions, el que ens permetràar escollir les proteïnes a analitzar. En general, s'observa que el suport per ESTs de les diferents prediccions no fou l'òptim, en molts casos només alguns dels exons són suportats per ESTs. Com ja s'ha dit aquest fet pot ser degut a que les llibreries d'ESTs siguin incompletes.
Així doncs, el procés simultani de comparar les prediccions gèniques dels diferents programes i la validació per ESTs ens permet seleccionar un total de 17 proteïnes (hem seleccionat aquelles proteïnes que són predites per diferents programes i/o validades per ESTs).
- A l'analitzar les diferents proteïnes amb el BLASTP observem que 8 de les proteïnes analitzades s'alineen correctament amb proteïnes Hox (elevat score i identitat i baix e-value): "Homeobox protein Hox A1", "Homeobox protein Hox A2", "Homeobox protein Hox A3", "Homeobox protein Hox A5", "Homeobox protein Hox A6", "Homeobox protein Hox A9", "Homeobox protein Hox A10", "Homeobox protein Hox A13". Aquestes proteïnes es troben agrupades en el genoma formant un cluster. Observem que tots els gens hox són en reverse, ja que com sabem presenten colinearitat espacial de 3' a 5'. D'altra banda també hem observat aliniament amb "EVX" (even-skipped, gen pair-rule de Drosophila).
També s'ha observat aliniaments amb proteïnes que pertanyen a d'altres cromosomes (proteïna ribosomal humana L7a, "High Mobility Group",TPM4, TPM3, "Histona Deacetilasa 4", "60S ribsomal protein L35", "matrix metalloproteinase 21"), de manera que podem concloure que algunes de les proteïnes predites contenen dominis similars a aquestes.
Continuant amb l'anàlisi de proteïnes, hem realitzat un aliniament de totes les proteïnes contra elles mateixes (amb el formatdb). Junt amb els resultats del BLASTP, podem concloure que gran part de les proteïnes s'alineen en diferents punts, el que ens fa suposar que s'alineen en l'homeodomini.
- Finalment si comparem els nostres resultats amb els del projecte ENCODE, observem que existeix bona correlació tot i que els gens "Hox A4", "Hox A7","Hox A11" no han estat predits en el nostre estudi.
Pàgines web:
- http://www.homeobox.cjb.net/
Referències bibliogràfiques:
- Hong et al. Structure and function of the HOX A1 human homeobox gene cDNA.Gene, 1995 Jul 4;159(2):209-14.
- Gilbert, Scott F.Developmental biology. 6a edició, Sunderland (Mass.) Sinauer Associates.
Agraïm a la resta de grups d'anàlisi de seqüències, la seva ajuda, comprensió, suport i paciència en aquestes últimes setmanes, sense vosaltres aquest treball no hagués estat possible.
També agraïm al nostre tutor, en Josep Abril, per quedar-se fins tard moltes tardes... Agraïm també l'ajuda d'altres professors en absència del nostre tutor, en especial, a en Robert Castelo.