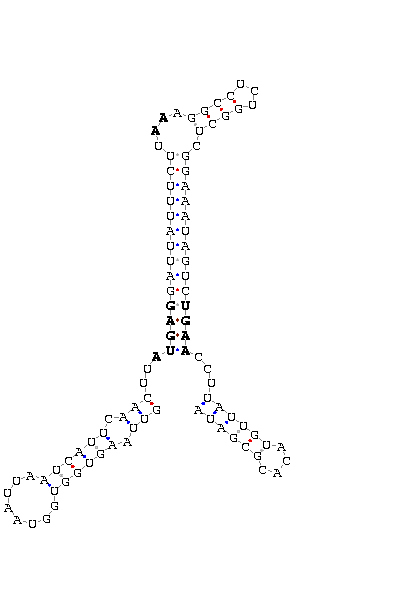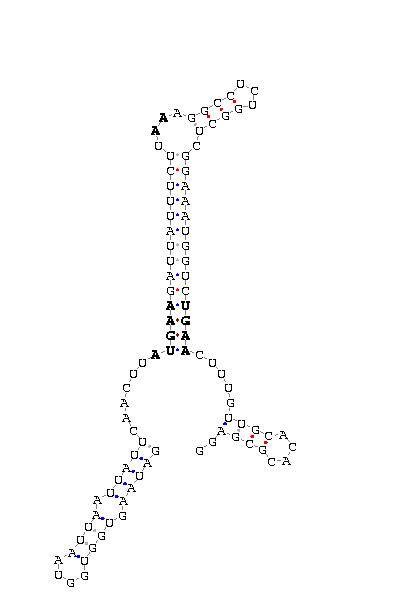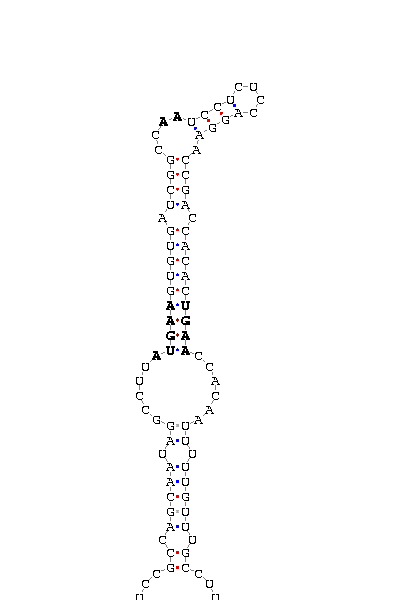The central dogma of the coding abc have changed
from the discovery of the 21st aminoacid, selenocysteine (Sec or U), coding from UGA codon,
usually stop codon. Bioinformatics have emerged as a powerful tool for the study of this coding bias.
The genome analysis programs available before this discovery were proned to incorrect annotation of sequences
containing UGA codons in-frame. It has been necessary the development of new programs that recognized
in-frame TGA code words as Sec in selenoprotein genes.
There have been identified selenoproteins from archea to eukarya, except in land plants
and yeast. The observation of a type of proteins that incorporate selenium suggested the important role of
selenium in cell metabolism and alimentation. The lack of selenium on the diet it has been related to
prostate cancer and other diseases. Selenium is a trace element that has been associated with cell-proliferation, cancer, development
and aging. This element occurs in animals primarly in selenoproteins. It could be important in the study
of the antioxidant cell function and redox balance. The selenocystein is placed in the catalytic domain,
suggesting its important role for the function of protein. It has been reported in mammals that mutations on some of the 25 selenoproteins
described lead to different disease. The number of selenoproteins differs depending on the taxonomic level, for example 3 in Drosophila
Selenoproteins are mainly RED-OX enzymes,such as glutathion peroxidase, thioredoxin
reductase and thyroid hormone deiodinases, and most of them are enzymes of their own production machinery.
The synthesis in eukaryotes is partially elucidated, but the knownledgement of selenoproteins in Drosophila.
is not yet completed.
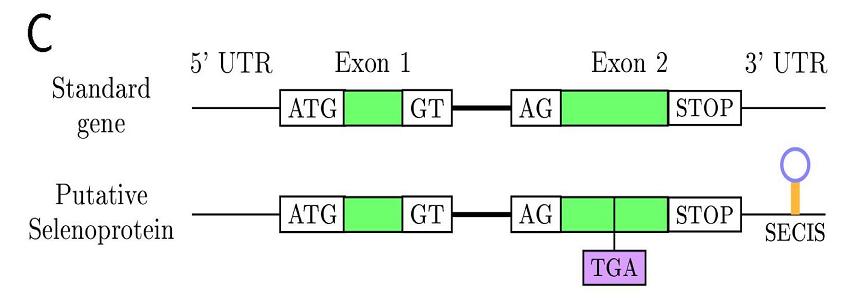
In eukaryotes this family of proteins are characterized for the SECIS element
(selenocystein insertion sequence), an mRNA motif placed in 3'UTR
allowing translational readthrough as selenocysteine. The SECIS-binding protein (SBP2) recognizes the SECIS
element for insertion of Sec into protein. Furthermore, a second factor, the specific Sec elongation protein (EFsec),
recognizes Sec-tRNA[Ser]Sec, and the resulting complex between EFsec-Sec-tRNA-SBP2 governs the donation of Sec
to the nascent selenopeptide in response to UGA codon. A nucleotid pattern sequence is conserved, but not the
secondary structure, that shows some degree of diversity between species.
Objectives
Nowadays we have been able to identify three selenoprotein genes
three selenoprotein genes in Drosophila melanogaster:
- Dsps2:
Selenophosphatase 2. It is a protein of 370 aminoacids coded by a gene composed for 3 exons.
The gene codes two different isoforms (A and B) by alternative splicing.
It's involved in selenocysteine biosynthesis by activating intracellular selenium, from selenide and ATP,
to form a selenium donor compound, selenophosphate.
- DselM(SelH)
BthD. It is a protein of 249 aminoacids coded by a gene located in chromosome X.
It's important for the selenium intake and probably involved in Drosophila life span.
There are two annotations of that protein, one with cysteine (Selenoprotein BthD precursor)
and another one with selenocysteine (CG11177-PA).
- DselG (SelK)
Glycine rich selenoprotein. It is 110 aminoacid protein coded by a gene located
in Chromosome X in tandem separed by only 300bp. There are two annotations of that protein,
one with Cysteine (G-rich selenoprotein) and
another one with selenocysteine (CG1844-PA). Its function remains unknown.
One of the aims of this project is to predict the characterized selenoproteins of
Drosophila melanogaster in other insecta species.
Other point of interest is to investigate in the homologue proteins if selenocysteine is conserved
along the species or if it is replaced by cysteine. It remains unknown the evolution pattern of cysteine and
selenocysteine in this family of proteins.
We expect to find all three homologues on other drosophilas due to the taxonomic similarity, but not
all of them on species further as Anopheles, Aedes and Apis. Those also found in
these species could indicated the importance of these proteins.
Go back
- Selenoproteins sequences
- Genomes sequences
Sequences from DroSpeGe: Drosophila Species Genomes, web site exclusive for Drosophila sequencing projects
- BLAST program: tBLASTn
Useful and suitable program that compares and aligns sequences.
Different programs can be run depending on the goal of our analysis.
We blasted a protein query sequence against nucleotide sequence database.
The program dynamically translated in all frames the query sequence. We use both versions,
the BLAST on-line and the command line. We run tBLASTn against insecta species, and another
one against arthropoda due to the different genomic data source. Not all the genomes of the organisms
that had to be analysed are placed together.
On-line BLAST version:
Command line BLAST:
export PATH=$PATH:/disc8/bin/:/disc8/bin/ncbiblast/:/disc8/soft/R/bin
export BLASTMAT="/disc8/bin/ncbiblast/data"
Using the commands formatdb
and blastall and creating
the text file .ncbirc
containing
[NCBI]
Data="/disc8/bin/ncbiblast/data"
Some of the target sequences are long contigs or even more, a whole shotgun containing whole
chromosomes due to the incomplete sequencing of these genomes.
In order to select the target sequence desired we export the program from persy server.
Fastachunk
.
That program is useful to cut sequences from a fasta input calling the following command:
fastachunk
Hits selection:
Acording to our goal of studying the evolution of selenoproteins two different
matches are interesting for us: Sec (U) - Sec (U) and Sec (U) - Cys (C). In order to asure the correct matches
we performed a handmade selection, instead of write down an script (we heard about the hard experience of
other groups trying to write an script to read BLAST output and get the sequences). Another criteria is the e-value obtained in BLAST, that we considered valid between 1e-05 and ~0.
We also discart those blast matches with a low score. We defined this rank after testing these sequences at Exonerate program and obtain empty outputs.
Gene Prediction
Using the command line version of programs from Persy server export PATH=$PATH:/disc8/bin
We first thought to use Genewise. We were stopped because the program has only two models of
prediction far from insecta: worm and human. That led to an error on the output (no hits found). Then, we choose
an equivalent program, Exonerate (version 0.8.2), that combines protein alignment to the genome and the prediction of
the gene structure with the best score in both strands based on Hidden Markov Models statistics.
In order to obtain an informative and pretty output we designed the following command:
exonerate -q protfilename -t DNAfilename -m protein2genome -W 3 --showtargetgff yes
where:
- -q: query (fasta format)
- -t: target (fasta format)
- -m: model
protein2genome: The query sequences were the characterized Drosophila melanogaster
protein blasted to a target DNA sequence.
- -w: Indicates the wordlength for protein words. By default it reads 6 by 6.
- showtargetgff: show also the output in gff format.
Looking for SECIS:
As we said before, Exonerate predict the 'exon-intron' structure in both strands.
Some of our outputs could be predicted on the negative strand. With those we need a pretreatment
in order to get the complementary strand from the target sequence.
That treatment will be useful to search the SECIS element directly, obtaining the
specific nucleotides on the target sequence (strand -) instead of the nucleotide positions on the father
sequence. We wrote down a command for the shell terminal to translate the target sequence to the reverse
and complementary strand (-):
FastaToTbl | rev | perl -lane '$F[0] =~ tr/ACGT/TGCA/; print "$F[1]\t$F[0]"' | TblToFasta
where:
- FastaToTbl: program that transform fasta format into table format of two columns, one for the
sequence ID and the other one containing the nucleotide sequence in one line.
- rev: reverse function: shown the sequence 3'-5' (and also the sequence ID).
- perl -lane: instead of writting an script we wrote a command line using perl language.
Each letter is refered to a perl command:
- l: enable line ending processing
- a: autosplit mode, combined with -n or -p (split $_ into @F)
- n: assume while(<>) {..}
- e: one line of program, omiting programfile
- $F[0] =~ tr/ACGT/TGCA/; print "$F[1]\t$F[0]": transliteration that permit to get the complementary
sequence.
- TblToFasta: from table format to fasta format.
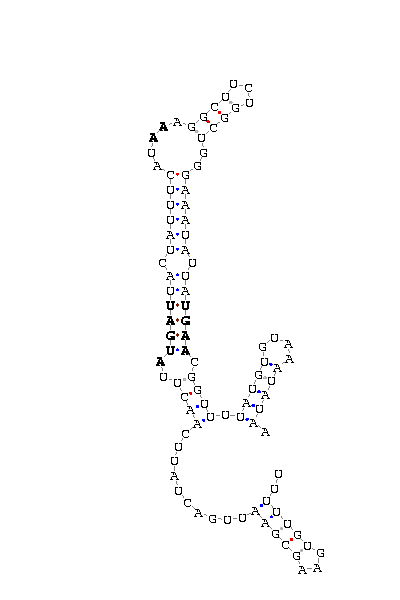
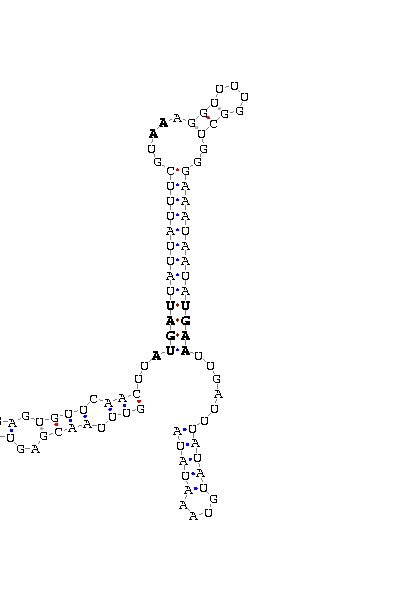
The search of possible SECIS elements was performed using one of SECISearch moduls
PATSCAN. That program identify
candidate of SECIS estructure from a nucleotide sequence (DNA) based on the conserved motifs.
It also predict the secondary structure of SECIS. A more accureted approach could be done using
Viena RNA package that combines the previous information with the free energy of the structure
resulting in a more complete prediction.
Selection of valid SECIS
First we search SECIS under default conditions (motif ATGA) and after that, if we didn't get any results
we tried the option canonical and non-canonical. We only took as useful those located in the 3'UTR, far
from the last exon roughly 1000 nucleotides.
Other programs used:
Go back
RESULTS
After running the programs needed for the analyse we show the results following this structure:
- - Name of organism
- - E-value (score) tBLASTn
- - Link to ALIGNMENT
- - Link to GFF format
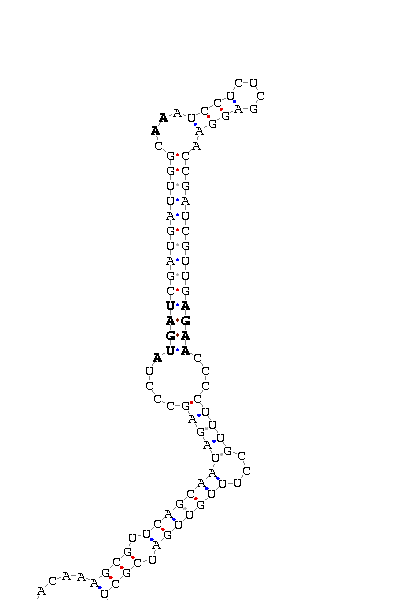
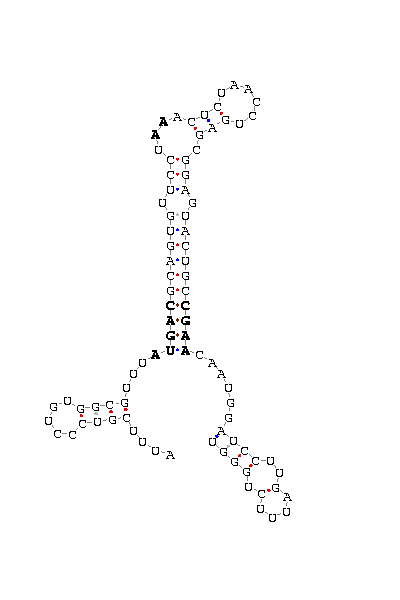
- - SECIS picture (only those placed in the valid rank)(See Methodolgy).
Drosophila melanogaster - Dsps2_D_mel.alignment Dsps2_D_mel.gff SECIS
Drosophila ananassae 1e-159 (316) Dsps2_D_ana.alignment Dsps2_D_ana.gff
Drosophila erecta 1e-134 (245) Dsps2_D_ere.alignment Dsps2_D_ere.gff
Drosophila mojavensis 9e-61 (168) Dsps2_D_moj.alignment Dsps2_D_moj.gff SECIS
Drosophila pseudoobscura 1e-146 (519)(2HITS) Dsps2_D_pse.alignment Dsps2_D_pse.gff
Drosophila simulans 0.0 (345) Dsps2_D_sim.alignment Dsps2_D_sim.gff SECIS
Drosophila virilis 1e-109 (182) Dsps2_D_vir.alignment Dsps2_D_vir.gff SECIS
Drosophila yakuba 0.0 (348) Dsps2_D_yak.alignment Dsps2_D_yak.gff SECIS
Anopheles gambiae 3e-82 (171) Dsps2_A_gam.alignment Dsps2_A_gam.gff
Drosophila melanogaster - DselM_D_mel.alignment DselM_D_mel.gff SECIS
Drosophila ananassae 1e-29 (130) DselM_D_ana.alignment DselM_D_ana.gff SECIS
Drosophila erecta 9e-62 (234) DselM_D_ere.alignment DselM_D_ere.gff
Drosophila pseudoobscura 1e-35 (150) DselM_D_pse.alignment DselM_D_pse.gff SECIS
Drosophila simulans 2e-66 (249) DselM_D_sim.alignment DselM_D_sim.gff
Drosophila virilis 4e-23 (107) DselM_D_vir.alignment DselM_D_vir.gff
Drosophila yakuba 5e-65 (213) DselM_D_yak.alignment DselM_D_yak.gff SECIS
Anopheles gambiae 3e-16 (86) (2 HITS) DselM_A_gam01.alignment DselM_A_gam01.gff
DselM_A_gam02.gff
Drosophila melanogaster - DselG_D_mel.alignment DselG_D_mel.gff SECIS
Drosophila ananassae 1e-16 (65) DselG_D_ana.alignment DselG_D_ana.gff SECIS
Drosophila simulans 4e-24 (110) DselG_D_sim.alignment DselG_D_sim.gff SECIS
Drosophila yakuba 8e-10 (56) DselG_D_yak.alignment DselG_D_yak.gff SECIS
Go back
DISCUSSION
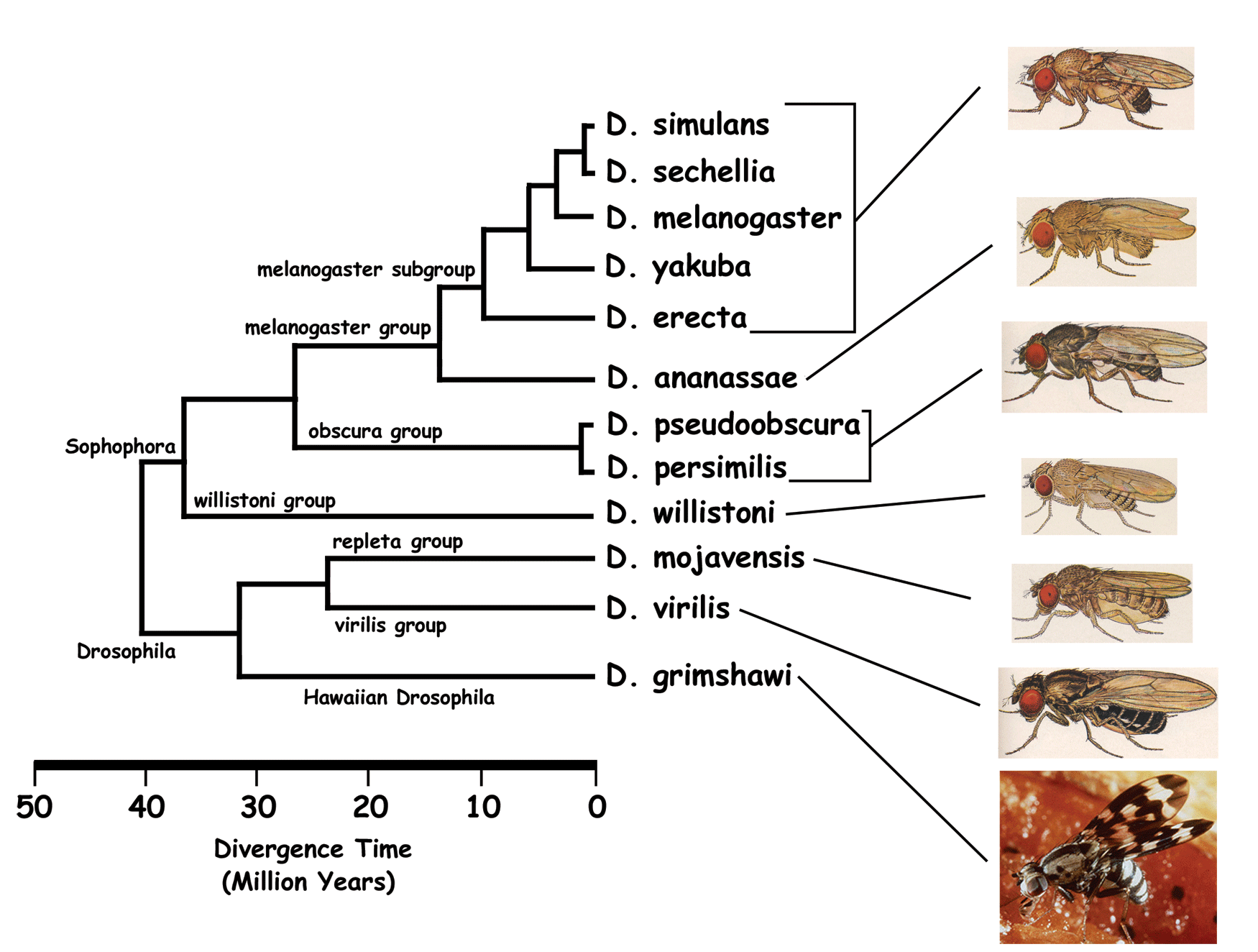
In order to analyse the evolutive differences of selenoproteins on Drosophila genus we used
the programs explained previously. Despite they were useful tools to observe the fingerprint
of evolution, we got several problems during the work that had to be solved to asure reliable results.
The first trouble was the incomplete sequencing of insecta genomes. Actually a lot of
research projects in Bioinformatics are focused on that issue. For our project it has been so helpful the web site
DroSpeGe. It includes a BLAST option link to almost all partially sequenced Drosophila genomes.
After running tBLASTn against all species, we did not get any hits for D. sechellia, D. willistoni, D. persimilis, D.grimshawi.
We were a little bit shocked, because we got in the same analyses hits for almost all other species. Surprisingly we found out
that there was no data underneath these genomes.
Another problem that we found along our way was the incorrect implementation of GFF output due to the split codon
We designed a program vulgar2gff able to read the vulgar output and write GFF format with the correct nucleotide annotation.
While we tried to set up the program and no results were obtained, we decided to change manually the annotation. Those presented on the
results are the correct ones.
In tBLASTn output we obtained some hits with U-cys matches and e-value around e-10/-15, but exonerate could not predict any gene structure from
those.
Primarly our study was not only focused on Drosophila species, but also other insecta such asApis mellifera, Aedes aegypti, Anopheles gambiae
In the results we only showed Anopheles gambiae because for the other organisms we could not find any hits (under our hit selection criteria)
or the hits found could not generate a prediction using Exonerate. We also discart some short alignments that could be random.
We discuss our results using two different criteria: one is the phylogenetic relationship between
Drosophila trying to find out if selenoproteins could help drawing a more likely phylogenetic tree.
And the other one, the differences on the protein sequence and gene structure among species that can lead to an accurated characteritzation of this gene,
analysing the most conserved motifs and the 'exon-intron' structure.
We start by following the selenoproteins evolution across Drosophila species. First of all
we generated a prediction of each protein using Exonerate with D.melanogaster protein query
and target genome. This output could work as guide for the validation of the predicted genes as homologous of the proteins.
- Drosophila yakuba
- This specie is the nearest to D.melanogaster and that is reflected on the complete alignment for the three proteins.
The raw score from Exonerate is so high and almost the same as D.melanogaster in all three proteins analysed.
The intronic structure shows is almost exactly, only dselM shows tiny differences on the length of the second intron.
No gaps were found on the alignments, only on the polyglycine tail of dselG. That does not
involved the functionality of that protein. SECIS element was predicted with a high score on the three proteins
- Drosophila simulans
- We found hit (U-TGA) on the three proteins, but the alignment is not as good as Yakuba (lower scores). For example,
the tail of DselG formed from one exon, was well aligned but it includes two stop codons before the U-TGA match. That fact
invalidate the alignment and also leads us to suspect that exonerate can not predict correctly
gene structures from proteins with special characteristics
such as polyglycine tail, as show in DselG. We predicted the complete structure of dselM and dsps2 and no gaps were observed.
The introns length are the same as those predicted in D.melanogaster. SECIS element was not found for DselM protein.
- Drosophila erecta
- Only dsps2 and DselM were predicted. Both show deletions on the DNA sequence, dselM (42 nucleotides)
and dsps2 (9 nucleotides). They also have shorter introns related
to D.melanogaster. Here we could notice the decrease of the raw score. The prediction of dsps2 is interrumped
because of the end of the contig. By the same problem, we were not able to look for SECIS element,
even though we try to join the consecutives contigs.
- Drosophila ananassae
- The three gene structures has been predicted, but with a lower score, in some cases
two times lower than D.melanogaster (e.g. DselM 384 vs 1182). The length of introns is
double than D.melanogaster. In DselM alignment we observed a shorter protein predicted,
due to an stop codon at 193 aminoacid position. However the SECIS element predicted was placed at 8621 nucleotide,
the end of stop codon. That could be explain by two hipothesis: is not an homologous protein, or it is a new selenoprotein
that could not be predicted using DselM as model on Exonerate.
- Drosophila pseudoobscura
- Only dselM and dsps2 could be predicted. We observed an strange structure on dsps2:
two different annotations for the same hit with no intronic sequence. That appeals us beacause
the prediction on the other Drosophila show four defined exons. We decide to analyse if this
structure could be pseudogen, by comparing the DNA sequence of pseudoobscura and yakuba
(CLUSTALW and BLAST2seq) to know if there was upstream and downstream (500bp)
conservation and also around the U-TGA match. We found that it was no conservation or not
good enough, thus we can conclude that is a pseudogen. Result.
DselM prediction shows a truncated protein of 124 aminoacids instead of 249 (D.mel). We first
asure if there was an stop codon after the 124th aminoacid. We saw next codon as codificant,
but not the following (126). That result agrees to the far distance between D.pseudoobscura
and D.melanogaster and is also supported by the robust SECIS found 200bp downstream on the 3'UTR.
- Drosophila mojavensis
- Only DselM and Dsps2 genes were predicted, but not as well as the others. The raw score
is not significative (276 vs 1182 in Dsps2 and 1136 vs 1836 in DselM). The difficulty of alignment
it is caused by the long distance between these species. The DselM predicted is show until the 116th aminoacid, but that does not
indicate that the protein is truncated. No stop codon was found and neither SECIS element. With these results we conclude that the
hit found was not correct or the annotation and sequencing incomplete. Further studies can solve this problem.
Dsps2 is predicted complete, with poor alignment, long introns and no high conservation around U-TGA match.
Also several gaps can be found. However, the SECIS search show one hit 14bp downstream.
- Drosophila virilis
- DselG is not predicted. Analysing DselM alignment we could notice that the prediction is
not correct for different reasons: shorter protein sequence aligned (128 versus 249),
no stop codon at the end of the alignment (300bp further), gaps on
DNA sequence and protein sequence too, low raw score and no SECIS found. On the other hand,
Dsps2 alignment show the same gene structure but differs on the conservation of selenocysteine
region, the intron length (four times) and some gaps more. The prediction of dsps2 gene and SECIS element is quite correct
and valid acording to our criteria.
- Anopheles gambiae
- Anopheles is the farest insecta that we got results. Only Dsps2 prediction shows
significative results. Otherwise an odd stop codon appears after the first intron, and an intron of 18260bp is predicted.
Analysing the sequence around we found that the donor and acceptor splicing sites were
not well predicted by exonerate. Probably it is the prediction with higher score, but not the
most related to the protein. This hipotesis is reforced by the SECIS element aprox. 100bp
further and the stop codon between them. DselM alignment could be random.