
ENr113
|
|
L'objectiu del nostre treball ha estat la caracterització de la regió anònima ENr113 del projecte ENCODE (ENCyclopedia Of DNA Elements). Hem començat obtenint la seqüència a partir de la base de dades de l'EMBL i a continuació hem emmascarat les repeticions.
Tot seguit hem realitzat prediccions de gens amb diferents programes i les hem validat llençant-les contra una base de dades de ESTs humans. En no trobar ESTs que suportin les nostres prediccions, hem relaxat els paràmetres per trobar resultats més significatius.
En el següent pas hem comparat les proteïnes que ens donaven les prediccions de gens amb una base de dades de proteïnes per intentar trobar homologies. Tan sols un dels gens predits presentava un domini conservat i hem intentat estudiar-lo.
Arribat aquest punt no hem pogut afirmar l'existència de cap gen conegut de manera que ens hem centrat en analitzar el per què d'aquest resultat.
La nostra seqüència correspon a un fragment de 500 kb que es troba en el braç llarg del cromosoma 4 (q26).
Obtenim la seqüència mitjançant la base de dades UCSC. Com que ens la faciliten en format FASTA la tabulem per caracteritzar-la.
Comencem calculant-ne la longitud amb la comanda:
awk '{print length($2)}' seq1.tbl
El resultat d'executar la comanda ens proporciona el resultat esperat: 500.000 pb.
A continuació determinem el contingut de C+G:
awk '{print $2}' seq1.tbl | fold -1 | sort | uniq -c | gawk '{print $2, $1/500000}'
El resultat que obtenim és el següent:
| A | 0.33118 |
| C | 0.172698 |
| G | 0.174488 |
| T | 0.321634 |
Per tant, el contingut C+G de la nostra seqüència és de 34.72%.
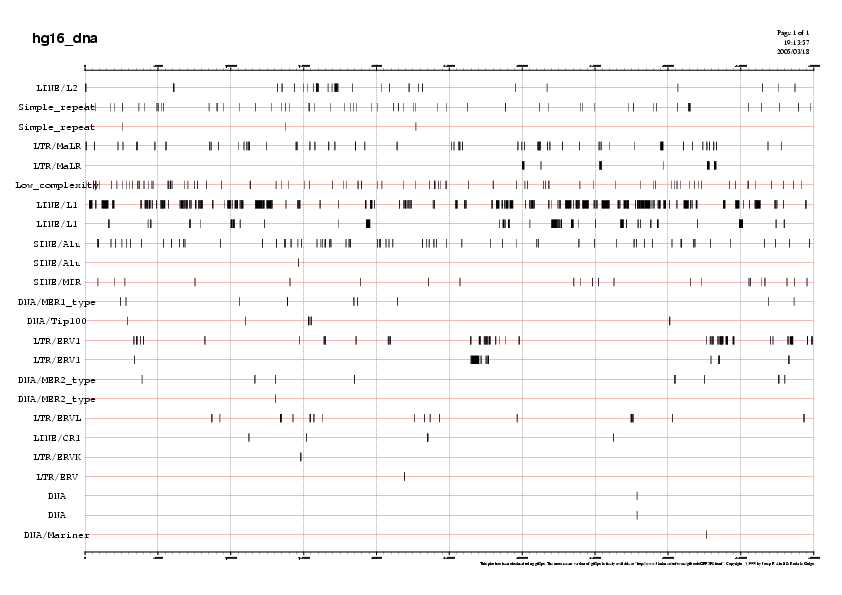
Tenint en compte que el DNA presenta gran quantitat d'elements repetitius que poden portar problemes a l'hora d'analitzar-los amb alguns programes, el que farem en primer lloc serà identificar aquestes repeticions i emmascarar-les per evitar el possible soroll.
Hi ha molts servidors web que permeten analitzar les repeticions, nosaltres hem utilitzat l' EMBL RepeatMasker server . El RepeatMasker et retorna tres arxius:
El primer arxiu, que conté la seqüència emmascarada, l'utilitzarem per córrer els programes de predicció de gens. En l'última taula podem observar que el 48.77% de la nostra regió està emmascarada i que és rica en LINEs.
Seria interessant veure la distribució de les repeticions al llarg de la seqüència. Ho farem utlitzant gff2ps, però abans caldrà convertir el fitxer al format gff:
Correm el gff2ps:
Ho passem a format png mitjançant la següent comanda:
Finalment, amb el kview observem:
grep hg16_dna repeatseq.out | \
awk 'BEGIN{ OFS="\t" }
{ print $5, $11, "repeat", $6, $7, ".", ".", "."; }
' > seq1out.gff
gff2ps seq1out.gff > seq1out.ps
convert -antialias -rotate 90 seq1out.ps seq1out.png
Existeixen diferents programes que realitzen prediccions de gens. Nosaltres n'hem utilitzat tres: el Geneid ( accedint via l'
IMIM geneid server) el Genscan (a través del MIT genscan server) i per últim el FGenesh.
Amb aquest programa vam córrer la seqüència emmascarada(repeatmaskedseq.fa) sencera.
Vam obtenir els resultats directament en gff (resultats_geneid.gff).
Aquests resultats són:
Geneid
| Gen | Inici | Final | Núm exons | Strand |
| 1 | 123350 | 150145 | 3 | Forward |
| 2 | 249598 | 250609 | 2 | Reverse |
| 3 | 304716 | 317724 | 2 | Reverse |
Vam tallar la seqüència emmascarada (repeatmaskedseq.fa) mitjançant fastachunk per tal de poder-la córrer en el genscan:
fastachunk repeatmaskedseq.fa 0 100000 | fold -60 > seq0a1.fa
fastachunk repeatmaskedseq.fa 90000 100000 | fold -60 > seq1a2.fa
fastachunk repeatmaskedseq.fa 190000 100000 | fold -60 > seq2a3.fa
fastachunk repeatmaskedseq.fa 290000 100000 | fold -60 > seq3a4.fa
fastachunk repeatmaskedseq.fa 390000 109950 | fold -60 > seq4a5_2.fa
Els trossos que vam obtenir els vam córrer amb el genscan obtenint els següents fitxers:
El següent pas va ser convertir-los a format gff. Ho varem fer a través de la comanda:
gawk 'BEGIN{OFS="\t"}
$2 ~ /Term|Intr|Init/ {
print "hg16_dna", "genscan", $2, start=($4<$5 ? $4 : $5),
end=($5<$4 ? $4 : $5), $13, $3, $7, $1;
}' seq0a1.txt | \
sed 's/\.[0-9][0-9]$//' > seq0a1.gff
Repetim la comanda per cada un dels fitxers resultants del genscan.
A continuació vam passar les coordenades a valors absoluts, ja que en haver tallat la seqüència en els diversos fragments haviem obtingut resultats relatius. Utilitzem la comanda:
gawk -v OFFSET=90000 '{$4=$4+OFFSET;$5=$5+OFFSET; print $0 }' seq1a2.gff > seq1a2_abs.gff
gawk -v OFFSET=190000 '{$4=$4+OFFSET;$5=$5+OFFSET; print $0 }' seq2a3.gff > seq2a3_abs.gff
gawk -v OFFSET=290000 '{$4=$4+OFFSET;$5=$5+OFFSET; print $0 }' seq3a4.gff > seq3a4_abs.gff
gawk -v OFFSET=390000 '{$4=$4+OFFSET;$5=$5+OFFSET; print $0 }' seq4a5_3.gff > seq4a5_3_abs.gff
Recordem que no cal modificar el primer fragment.
El següent pas consistirà en enumerar els diferents fragments segons el gen i l'exó que hagin predit:
gawk 'BEGIN{OFS="\t"}
{print "hg_16_2", $2,$3, $4, $5, $6, $7, $8, "Seq2_"$9"";
}' seq1a2_abs.gff > seq1a2_trossets.gff
Un cop tenim els fragments en valors absoluts i enumerats els concatenem. Caldrà executar una comanda per tal de descartar els exons repetits a causa de la superposició. Ens quedarem amb aquells d'score més elevat o que no trenquin el marc de lectura.
cat seq0a1_trosets.gff seq1a2_trossets.gff seq2a3_trossets.gff seq3a4_trossets.gff seq4a5_3_trossets.gff > seq0a5_cat.gff
En observar el fitxer resultant (seq0a5_cat.gff) veiem que el primer fragment i el segon tenen un gen superposat. Eliminen un dels dos ja que es tracta del mateix gen (té les mateixes coordenades i score) obtenint el fitxer seq0a5cat.gff.
Els resultats són els següents:
| Gen | Inici | Final | Núm exons | Strand |
| 1 | 94060 | 96756 | 3 | Forward |
| 2 | 110228 | 141085 | 2 | Reverse |
| 3 | 249598 | 250609 | 2 | Reverse |
| 4 | 340730 | 347022 | 2 | Reverse |
| 5 | 481414 | 496367 | 2 | Reverse |
Aquest programa ens permet córrer la seqüència completa i obtenim el document ( fgenesh.txt ).
Hem de convertir el fitxer a format gff i ho fem mitjançant un programa que ens permeti posar el frame:
#!/bin/gawk -f
BEGIN {
SEQ = ARGV[1];
ARGV[1] = "";
}
$4 ~ /^CDS/ {
if ($2 == "+") {
if ($4 == "CDSf") {
L = 0;
};
l = $7 - $5 + 1;
F = (3 - (L % 3)) % 3;
print SEQ,"FGENESH",$4,$5,$7,$8,$2,F,$1;
#,($4 == "CDSl" ? "## "(L+l)%3 : "");
L = L + l;
} else {
if ($4 == "CDSl") {
L = 0;
};
l = $7 - $5 + 1;
L = L + l;
F = L % 3;
print SEQ,"FGENESH",$4,$5,$7,$8,$2,F,$1;
#,($4 == "CDSf" ? "##" : "");
};
}
I executem la següent comanda:
./fgenesh2gff.awk "hg16_dna" genesh.txt > fgenesh.gff
Els resultats de la predicció per FGenesh són:
| Gen | Inici | Final | Núm exons | Strand |
| 1 | 62256 | 96758 | 2 | Forward |
| 2 | 250040 | 250594 | 1 | Reverse |
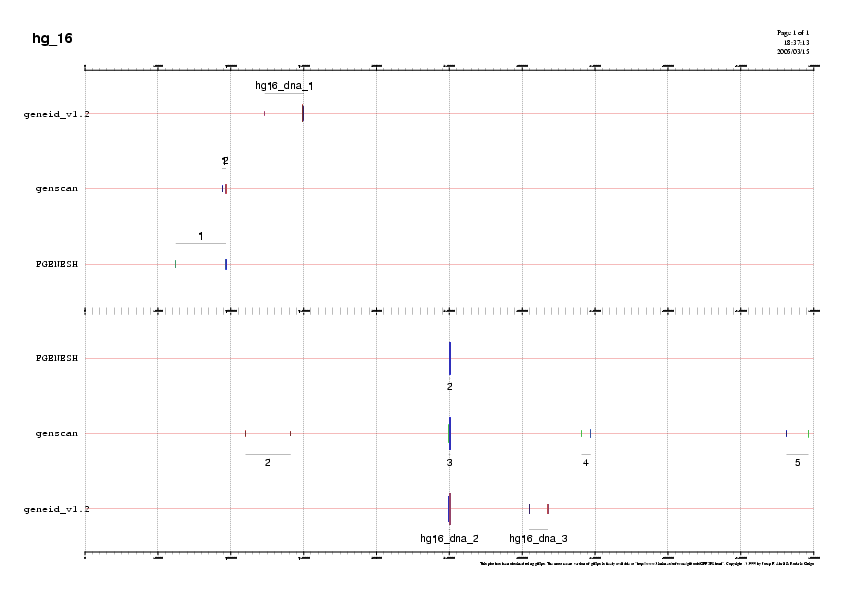
Arribat aquest punt tots els resultats dels diferents programes de prediccions de gens es troben en format gff i les dades en valors absoluts. Seria interessant obtenir un gràfic que ens permeti veure alhora les tres prediccions. Per fer-ho crearem un fitxer que contindrà totes les dades:
gawk '$1 !~ /^\#/ {$1="hg_16"; print $0}' geneid.gff seq0a5cat.gff genesh.gff > allinone.gff
A partir d'aquest fitxer farem el gràfic amb gff2ps:
gff2ps allinone.gff > prediccions3.ps
convert -rotate 90 prediccions3.ps prediccions3.png
La representació obtinguda és la següent:
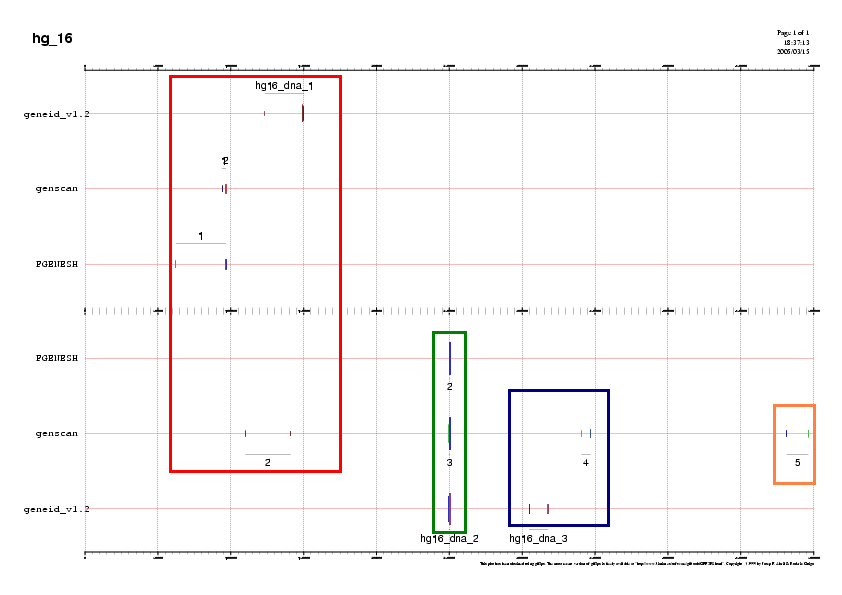
El primer que vam fer va ser dividir la seqüència en quatre regions (que corresponen a la localització dels possibles gens). Vam determinar-ne les coordenades en un emacs ( regions.tbl) i tot seguit vam executar la següent comanda:
Obtenim:
En la propera figura veiem aquestes regions sobre el gràfic:
egrep -v '^\#' regions.tbl | while read SEQ INI END; do { LEN=`expr $END - $INI + 1`; echo "#--> "$SEQ"--->"$INI"<-->"$LEN"<--";
( echo ">"$SEQ"."$INI"-"$LEN ; fastachunk repeatmaskedseq.fa $INI $LEN | fold -60 ) > regions.$SEQ.fa; }; done
regions.reg1.fa
regions.reg2.fa
regions.reg3.fa
regions.reg4.fa
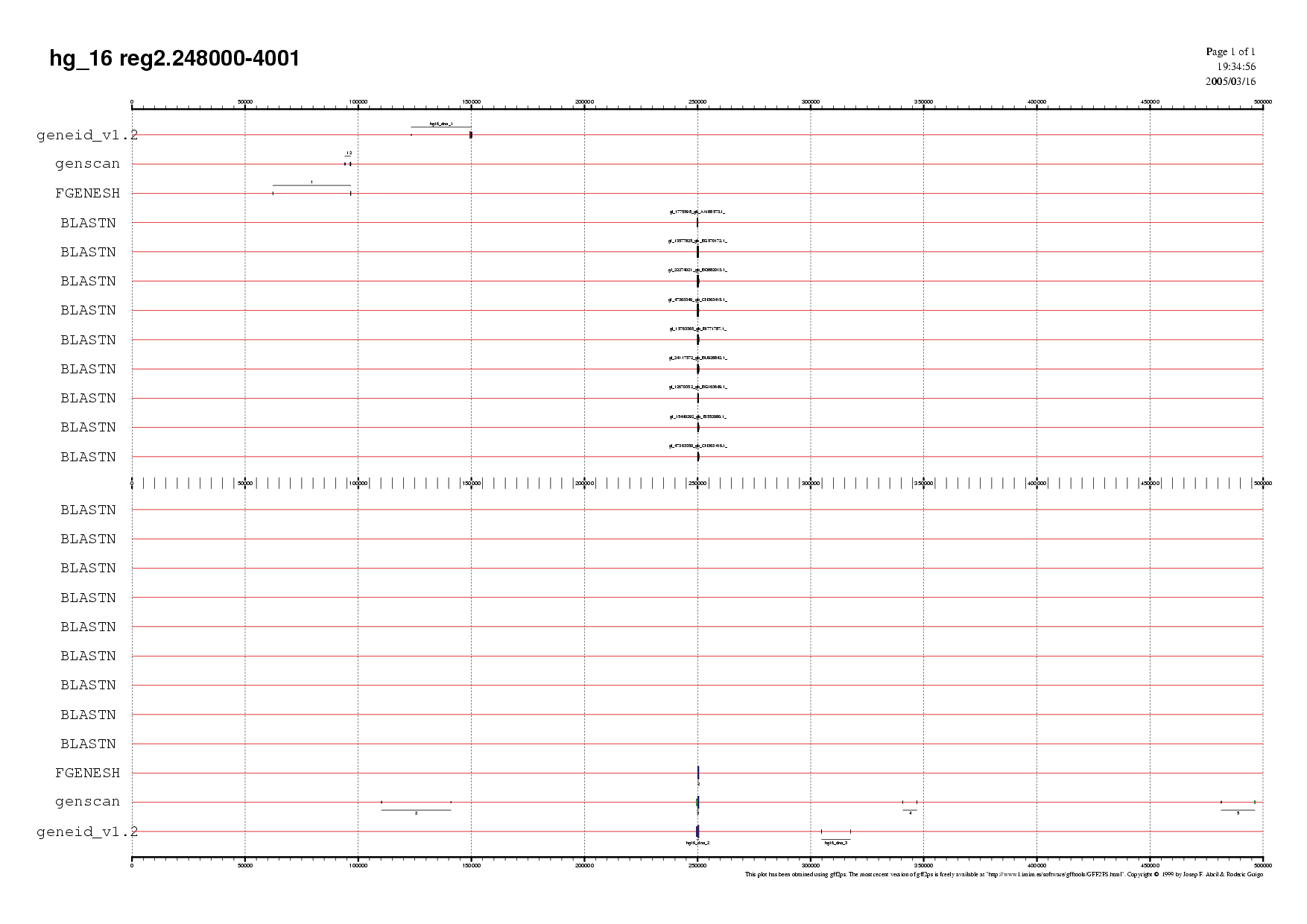
Tan bon punt aconseguides les regions vam passar a córrer el Megablast amb una base de dades d'ESTs humans. Els ESTs (Expressed Sequence Tags) són petits fragments de DNA (200-500 nucleòtids) generats per la seqüenciació d'un o dels dos extrems d'un gen (5' i 3'). Poden ser utilitzats per identificar gens desconeguts i validar els resultats dels programes de predicció de gens.
En un primer moment vam fer-ho amb els paràmetres pre-establerts del megablast però en fer una primera valoració veiem que els resultats no eren satisfactoris perquè no trobavem ESTs que suportessin més d'un exó. Vam decidir relaxar els paràmetres amb la intenció d'aconseguir resultats que poguessin validar alguna de les nostres prediccions.
Els paràmetres comuns a totes les regions varen ser els següents:
Options
Format
Regió 1: vam córrer diferents megablasts identificant l'entrada com humà, primats, mamífers i vertebrats. No veiem millores en els resultats en relaxar els paràmetres, per tant vam quedar-nos amb els obtinguts fixant humà com entrada. Vam guardar-los com reg1est.blast.txt.
Regió 2: en aquest cas obteniem una millora en els resultats en fixar mamífers com a entrada (reg2mest.blast.txt).
Regió 3: ens quedem amb els resultats del megablast que té humà per entrada (reg3est.blast.txt).
Regió 4: també ens quedem amb els resultats del megablast que té humà per entrada (reg4est.blast.txt).
Tot seguit hem de transformar els fitxers que hem obtingut a format gff. Ho farem utilitzant parseblast:
parseblast.pl -G reg1est.blast.txt > reg1est.blast.gff
parseblast.pl -G reg2mest.blast.txt > reg2mest.blast.gff
parseblast.pl -G reg3est.blast.txt > reg3est.blast.gff
parseblast.pl -G reg4est.blast.txt > reg4est.blast.gff
Un cop els fitxers es troben en format gff, passem les coordenades d'inici i final a valors absoluts:
gawk 'BEGIN{OFS="\t"}{$4=$4+61000; $5=$5+61000;print}' reg1est.blast.gff > reg1est.blast.abs.gff
gawk 'BEGIN{OFS="\t"}{$4=$4+248000; $5=$5+248000;print}' reg2mest.blast.gff > reg2mest.blast.abs.gff
gawk 'BEGIN{OFS="\t"}{$4=$4+303000; $5=$5+303000;print}' reg3est.blast.gff > reg3est.blast.abs.gff
gawk 'BEGIN{OFS="\t"}{$4=$4+480000; $5=$5+480000;print}' reg4est.blast.gff > reg4est.blast.abs.gff
A continuació tallem i ordenem la columna 9, per veure les seqüències EST que ens validen més d'un exó. Utilitzem la següent comanda:
gawk '{ print $9 }' reg1est.blast.abs.gff | sort | uniq -c | sort -nr | more
gawk '{ print $9 }' reg2mest.blast.abs.gff | sort | uniq -c | sort -nr | more
gawk '{ print $9 }' reg3est.blast.abs.gff | sort | uniq -c | sort -nr | more
gawk '{ print $9 }' reg4est.blast.abs.gff | sort | uniq -c | sort -nr | more
L'única regió validada per EST amb més d'un exó ha estat la regió 2. En les altres regions no han sortit evidències, tot i que hem provat de canviar les coordenades comparant-ho amb diferents bases de dades les sequencies d'EST només prediuen un exó i no l'estructura que hem observat en "prediccions3.png". Pot ser que aquests gens no s'expressin gaire o tan sols en fases molt concretes del cicle cel.lular i que no estiguin en la base de dades.
En tot cas, seguim l'anàlisi del resultat concatenant les regions en valors absoluts per poder veure el resultat en un gràfic juntament amb les prediccions:
cat reg1est.blast.abs.gff reg2mest.blast.abs.gff reg3est.blast.abs.gff reg4est.blast.abs.gff > reg1a4est.blast.gff
De la concatenada tan sols ens interessen les seqüències que continguin llocs de splicing (només hi trobarem en la regió 2). El programa awk "getsplicedhsp.awk", ens permet quedar-nos amb aquestes seqüències:
gawk -f getsplicedhsp.awk reg1a4est.blast.gff > reg1a4est.spliced.gff
Per observar-ho en un gràfic utilitzem gff2ps:
gff2ps allinone.gff reg1a4est.spliced.gff > est+prediccions3.ps
convert -antialias -rotate 90 -density 150 est+prediccions3.ps est+prediccions3.png
Decidim fer un "zoom" de "est+prediccions3.png" en la regió 2 (la que ens interessa) per veure si la validació d'EST coincideix amb els 2 exons que ens han predit els programes:
gff2ps allinone.gff reg1a4est.spliced.gff > est+prediccions3_zoom.ps
convert -rotate 90 est+prediccions3_zoom.ps est+prediccions3_zoom.png
La representació que hem obtingut ha estat:
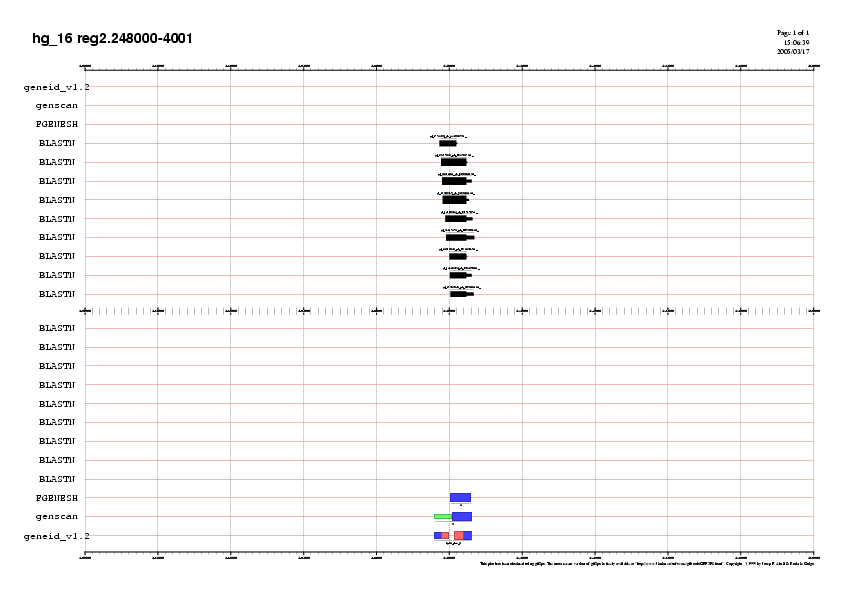
En veure el gràfic observem que els EST es troben en realitat solapats, és a dir, no hi ha espai de splicing entre ells, es tracta de dues prediccions diferents on una té més score que l'altra i acaben solapant. No és el resultat que esperavem ja que no ens permet validar la nostra predicció.
Tot i que no hem pogut validar cap de les prediccions per EST, seguirem analitzant la nostra seqüència i ho farem a través de les proteïnes que s'obteniem segons els programes de prediccions de gens.
La següent taula mostra la seqüència d'aminoàcids de les proteïnes predites segons cada programa:
| Geneid | |
| Genscan | |
| FGenesh |
Varem utilitzar el blastp per tal de trobar homologies utilitzant la base de dades del NCBI (All non-redundant GenBank CDS translations+PDB+SwissProt+PIR+PRF). Igual que en la cerca d'ESTs ha estat necessari modificar els paràmetres per tal de trobar resultats significatius. Bàsicament varem modificar l'apartat d'opcions, on vam seleccionar diferents organismes i ens vam quedar amb els millors resultats.
Després d'efectuar tots aquests aliniaments canviant els paràmetres vam veure que relaxar-los no permetia arribar a uns resultats millors: teniem més aliniaments, però l'score era molt baix.
Finalment només hem trobat homologia significativa en la regió 2 i per tant decidim estudiar-la més a fons. Si ens fixem en l'aliniament amb la proteïna UMPH1 que ens proposa el blastp (aliniamentUMPH1.html) veiem una part de la regió que no s'alinia amb aquesta. Pensem que pot tractar-se d'un exó que no han predit els programes de predicció de gens o bé d'un tros de seqüència que s'ha perdut al llarg de l'evolució. Per comprovar-ho fem un blast de la nostra seqüència (nucleòtids) contra UMPH1 (hipotetical.html). Fem els següents passos:
1. Fem un fastachunk per seleccionar la regió 2 a partir de la repeatmaskedseq:
fastachunk repeatmaskedseq.fa 240000 15000 | fold -60 > reg2hipo.fa
2. Fem formatdb de la nostra seqüència:
formatdb -p F -i reg2hipo.fa
3. Executem la següent comanda per fer el blast:
blastall -p tblastn -d reg2hipo.fa -i hipoteticalprot.fa -e 0.1 > reg2+hipo_blast.out
4. Ho passem a format gff:
parseblast.pl -S -G reg2+hipo_blast.out | sort > reg2+hipo_blast.gff
En analitzar els resultats obtinguts observem que les nostres hipòtesis no eren certes, ja que les seqüències es troben en sentits oposats.
Després de dur a terme tots els anàlisis ja citats la primera conclusió a la que arribem és que no podem afirmar amb seguretat l'existència de cap gen en la nostra seqüència.
Aquest resultat era d'esperar: un dels primers passos ha estat la determinació del contingut C+G i ens ha donat que és del 34.72%, massa baix per tractar-se d'una regió codificant perquè en aquestes trobem,normalment alts continguts de C+G.
Els resultats dels programes de predicció de gens (Geneid, Genscan i FGenesh) seguien la mateixa direcció: els gens predits eren pocs. Tot i així semblava que en la regió 2 podriem trobar-hi un gen perquè els tres programes coincidien indicant-ne la presència (els resultats del Geneid i Genscan predien que el gen tenia 2 exons, mentre que el FGenesh predia tan sols un exó).
La validació per ESTs va seguir la línia marcada pels resultats de la predicció de gens: en general no hi havia ESTs que suportessin més d'un exó, ni tan sols relaxant els paràmetres. Vam realitzar molts Blasts modificant els diferents paràmetres (mida de la paraula, número d'aliniaments que ens mostra el programa...) per intentar trobar informació rellevant. L'única regió on vam trobar resultats una mica més esperançadors va ser altra vegada la 2. Vam decidir aprofundir en aquesta regió i vam fer un zoom: no es tractava d'un EST que estés suportant més d'un exó, sinó de dues puntuacions diferents d'un mateix EST que s'estavem aliniant de manera superposada. Així, doncs, no hi havia ESTs suportant el gen de dos exons que havien predit el Geneid i el Genscan.
En mirar homologia amb proteïnes també vam haver de relaxar els paràmetres, ja que en un principi tampoc trobavem resultats positius. De totes maneres cal tenir en compte que si relaxes massa els paràmetres els resultats no són bons: trobes aliniaments molt curts i amb un score baix. Vam arribar a la conclusió que si no trobes resultats comparant amb humà o primats no val la pena provar-ho amb altres organismes.
En fer la cerca amb el Swissprot vam trobar un domini conservat en la regió 2: s'aliniava amb la proteïna UMPH1 (Uridine Monophosphate Hydrolase-1), però hi havia una part que no s'aliniava. Era d'un tros prou gran com per pensar que podria tractar-se d'un exó que, o bé no havia estat predit pels programes de predicció de gens o podia haver-se perdut al llarg de l'evolució. Per estudiar-la més a fons vam aliniar la part corresponent de la regió 2 contra UMPH-1 (vam establir la nostra seqüència com a bases de dades i la UMPH-1 com a 'query'). Vam observar que en l'aliniament cada proteïna anava en un sentit diferent; amb la qual cosa era molt poc probable que es tractés de la mateixa proteïna.
Arribat aquest punt ens plantegem perquè no hem trobat resultats positius.Observem un gràfic que descriu la nostra seqüència (dades proporcionades pel UCSC Genome Browser on Human July 2003 Assembly).
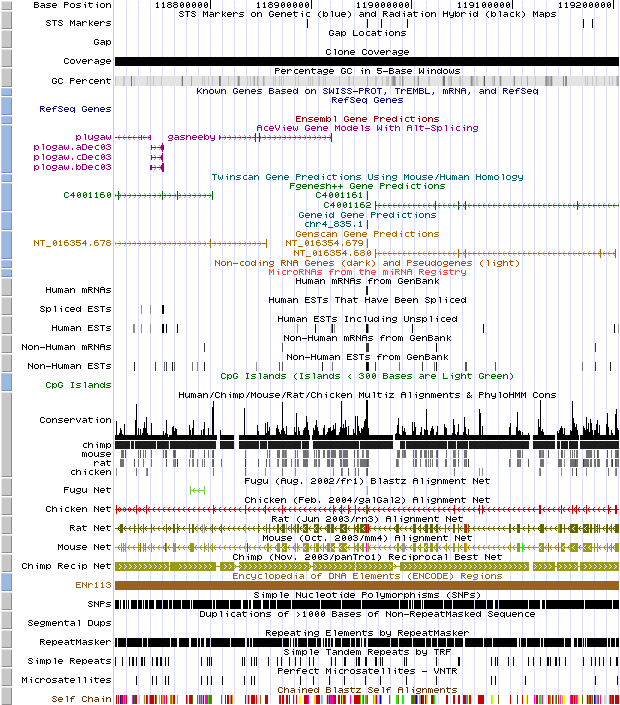
Pensem que potser ha estat perquè estavem treballant amb una part del genoma encara no seqüenciada, però veiem que no és així perquè té una cobertura alta.
Sen's confirma el baix contingut de C+G (el gràfic és d'un gris molt clar que indica aquest baix contingut).
Pel que fa a les bases de dades Swissprot, TrEMBL... veiem que no coneixen gens en aquesta seqüència.
En aquest gràfic també hi apareixen les prediccions de diversos programes de predicció de gens:
És interessant observar que en alguns casos no trobem l'inici del gen. Això podria explicar perquè les nostres prediccions no són gaire bones, ja que el fet de no trobar l'exó d'inici pot comportar problemes al programa a l'hora de trobar la resta d'exons o de classificar-los com a inici, intermig o terminal.
Totes les prediccions coincideixen situant un gen a la meitat de la seqüència; amb el qual també coincideixen les nostres prediccions.
Seria possible que estéssim treballant en una seqüència rica en RNAs no codificants i per això no trobem homologia en la base de dades de proteïnes. També podria tractar-se d'una regió rica en microRNAs: gens molt curts i antisense. Deduïm que aquest no és el cas, perquè en el gràfic veiem que la nostra seqüència no codifica per aquests tipus de RNAs ni tampoc en pseudogens.
En la part corresponent a 'Spliced ESTs' no hi trobem aquell gen situat a la part central de la seqüència, ja que, tal i com haviem vist, encara que semblés que podia tenir splicing en realitat no era un mateix EST suportant dos exons, sinó que eren dos possibles aliniaments. Veiem que apareix un 'Spliced EST' en una zona on nostaltres no n'hi haviem detectat. En canvi si que veiem l'EST no 'spliceat' que haviem trobat al mig de la seqüència.
Veiem que no hi ha illes CpG. Sovint les trobem en la regió 5' de gens de vertebrats. Aquest fet reafirma encara més els nostres resultats pel que fa a l'absència de gens.
En aquest gràfic també apareix homologia amb altres espècies (rat, mouse, ximpanze, fugu). És normal que no trobéssim homologia perquè en tractar-se d'una regió no codificant no es troba sota pressió selectiva i admet més canvis que una seqüència codificant. Per això en relaxar els paràmetres per intentar trobar ortòlegs no trobavem res. No hi ha conservació d'aquesta seqüència entre les diferents espècies.
Aquest fet es reforça si ens fixem en els SNPs: veiem que n'hi ha molts, cosa que implica diferències entre les seqüències que s'han analizat. Hi ha polimorfisme perquè no sembla ser una regió que tingui una funció imprescindible.
Observem que hi ha més conservació amb el ximpanzé perquè es tracta d'una espècie filogenèticament propera a la nostra i com que la divergència és relativament propera no hi ha hagut temps perquè es produïssin grans canvis. Podem observar-ho en el gràfic de conservació de color negre i també en les caixetes de les 'nets' de cada espècie. Veiem que hi ha molt pocs pics en la conservació i uns descensos bruscs i sobtats.
En la barra del Repeat Masker s'aprecia que molta de la seqüència està emmascarada (fet que coincideix amb els resultats que vam obtenir).
Pel que fa al gràfic de 'Self Chain' veiem que predomina el color grana, que correspon al cromosoma 4 (cromosoma on es troba la seqüència amb la que hem estat treballant). Si observéssim una porció llarga d'un altre color podria indicar que hi ha hagut recombinació amb un altre cromosoma.
Arribat aquest punt i havent comparat els nostres resultats amb les dades proporcionades per UCSC Genome Browser, tan sols queda preguntar-nos qué haguéssim pogut fer per millorar-los. Creiem que ampliant la nostra seqüència per davant i per darrera i sometent-la als programes de predicció de gens possiblement els resultats obtinguts s'haguéssin assemblat més als de l'UCSC Genome Browser perquè, tal com hem dit, ens manquen els inicis dels gens (segons les prediccions d'UCSC), cosa que perjudica la predicció.
No ens n'haguéssim sortit sense l'ajuda i paciència de:
GRÀCIES A TOTS!!! Laia i Montse
Agraïments