
In order to obtain our query sequences, we generated a phylogenetic tree which contains all the species found in the data base SelenoDB with the aim to identify the species with the closest phylogenetic relationship with Vipera berus. The phylogenetic tree was generated with PhyloT software, which is based on NCBI taxonomy (available online at: http://phylot.biobyte.de/) and we visualized it using Interactive Tree Of Life (iTOL) software[12][13].
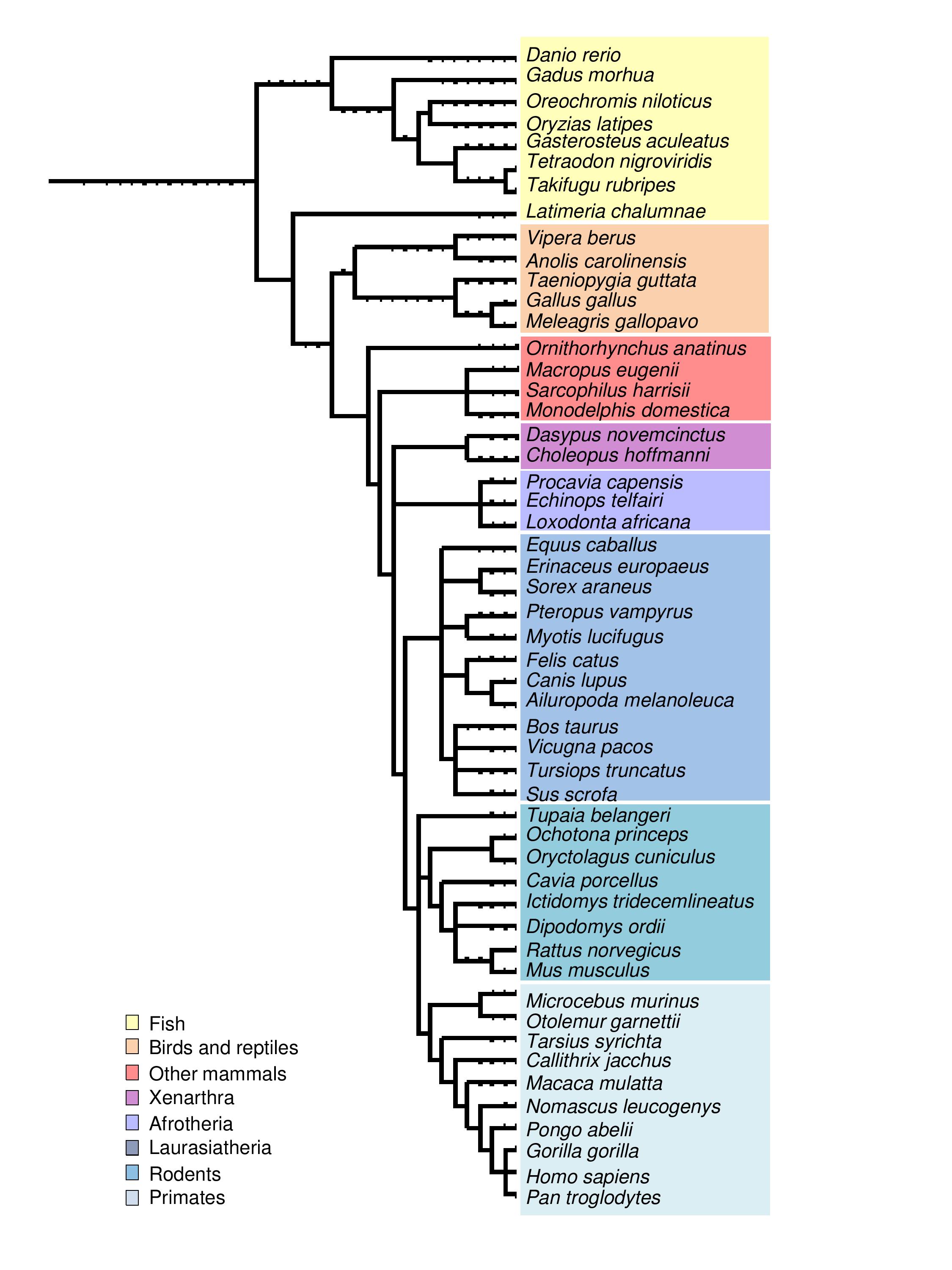
Figure 1. Phylogentic tree containing all SelenoDB species and Vipera berus.
As Figure 1 shows, the closest species to Vipera berus in SelenoDB is Anolis carolinensis. Therefore, we used its selenoprotein sequences as querys. These proteins were divided in red proteins (selenocystein contained in their sequence) and green proteins (cystein homologous and essential proteins of selenoprotein synthesis).
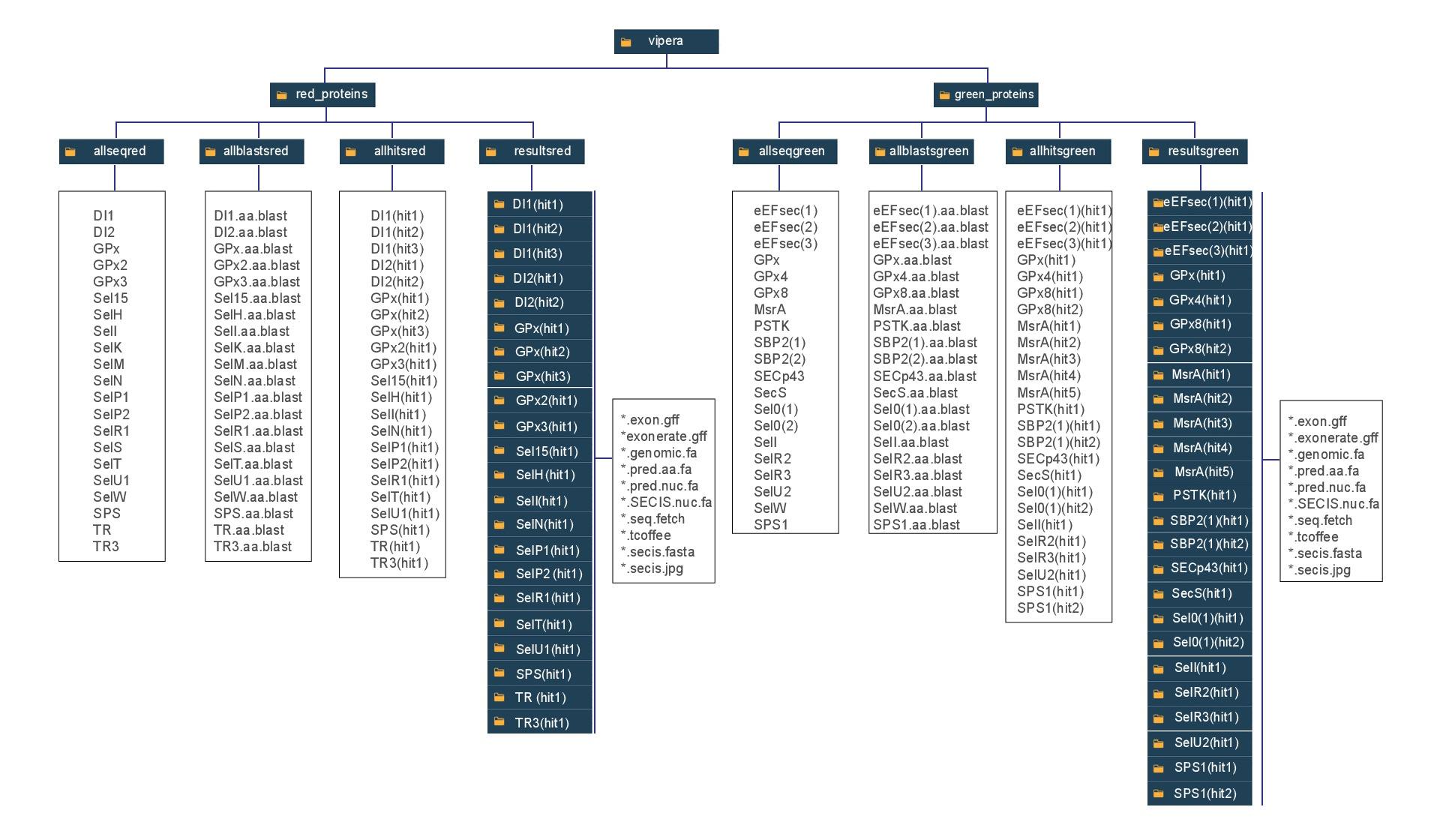
Click here to visualize the folder's system used in this project, where the folders are referred as black boxes and text files as whites boxes. This is specially important to understand the paths used in the scripts.
Once the Anolis carolinensis querys were obtained, we changed the U (selenocystein) for an X in the red proteins sequence to avoid errors during the process (script in bash).
A tblastn was runned with Anolis carolinensisquerys against the Vipera berus genome. tblastn modality takes an aminoacid sequence as a query and compares it to the reference genome , which is translated to its 6 possible frames. We automatized this process in a bash script, that allowed us to obtain the blast output for each protein (red_proteins script and green_proteins script). Click on the following links to find the sripts: red_proteins and green_proteins.
When the blast outputs were obtained, we analyzed them manually. The manual selection of the best hits allowed us to take into account different parameters and not simply set a threshold E-value. Hence, we followed the next steps to select the hits:
1. Selection of the best hits and discard of the worse. We payed attention to the E-value, which indicates how many times do we expect such a good alignment by chance: lower the E-value, more unlikely it will be to find the hit by chance. We kept all the hits with an E-value lower than 0.0001.
2. Alignments. We analyzed the following parameters:
2.1. E-value: Multiple hits can be found in the same scaffold, each of them with its value. If one E-value was high, we discarded it.
2.2. Selenocystein alignment (U): it is important that the U of the query aligns with a STOP codon (* symbol) or a Cys.
2.3. Alignment overlap: our hypothesis is that the hits from the same sequence correspond to different exons of the same gene and, as a consequence, it is expected that the last aligned aminoacid coincides with the first one of the next hit, with minimal overlap. Moreover, we took into account that the distance among the alignments must correspond to the intron length in Vipera berus genome. As Vipera berus genomes has not been annotated yet, we used the median intron length of a coding transcript in Anolis carolinensis, which is 5041 bp (min: 9bp; max: 856457 bp)[14].
2.4. Frame: in those hits from the same scaffold, we also took into account that the frame was the same (positive or negative). Moreover, the start and end positions depend on the frame.
3. When a hit fulfilled our criteria, we put its contig's ID, start and end in a new text file, which we used to extract the region of interest.
The rest of the process to search selenoproteins in Vipera berus genome was done automatically with a bash script for the selenoproteins (red_proteins) and the homologous and machinery (green_proteins).
Once the hits were selected, the regions of interest have to be extracted from the Vipera berus genome. First, the sequence (scaffold) which contain the region must be obtained using fastafetch:
Then, the region has to be extracted from the output sequence of fastafetch, as fastasubseq only works on a fasta file containing one sequence:
Start and length estimation: the start and end of the hit have to be extended 50,000 nucleotides per site, in order to obtain a region of interest which contains all the gene, always taking into account that the start must be higher than 1 and the length lower of the contig's length (lengths.fa file).
From the region of interest of each selected hit, we proceed to predict the gene structure:
- m p2g: protein to gene mode
- showtargetgff: outout in GFF format
- q: query (Anolis carolinensisprotein)
- t: target (Vipera berus region of interest)
Moreover, we extracted only the exons predictions from the whole gene structure:
Once we obtained the gff file, it has to be converted to fasta format using the perl program fastaseqfromGFF.pl:
Finally, the predicted sequence was translated from cDNA to protein:
The -F option indicates 'forward' and the nucleotide position where the traduction will began. As the protein prediction is done from cDNA (exons), we used the option -F 1.
When the fastatraslate finds a stop codon TGA, it writes a *, which may supose a wrong tcoffee alignment. For this reason, we changed the * for X, so we can identify an alignment between the selenocystein of the query (also changed to X at the beginnig of the process) with a TGA or a Cystein of the predicted protein.
When we obtained the predicted aminoacid sequence, we compared it with the query of Anolis carolinensis from the same protein, by aligning them using tcoffee.
Concerning to the homologous proteins, a previous alignment must be performed with its selenoproteins in order to identify in which position is localted the cystein. This can allow us to detect if it aligns with our predicted sequence in the tcoffee output.
In order to obtain the absolute coordenates in relation to the contig of the predicted genes and exons, we generated a script to automatize the calculation: fastasubseq start + relative position obtained in exonerate's output ($sample.exonerate.gff). Thus, we get the absolute positions of the genes as well as of each of the predicted exons. In these links, you can find the scripts: coordenades_gens_absolutes.sh and coordenades_exons_absolutes.sh for the red_proteins, coordenades_gensgreen_absolutes.sh and coordenades_exonsgreen_absolutes.sh for the green_proteins.
Finally, we annotate in which aminoacid position of the predicted protein is located the aligned Selenocystein or Cystein.
SrciSearch as a tool for prediction of eukaryotic SECIs elements. It is an open software available online at: http://gladyshevlab.org/SelenoproteinPredictionServer/ [15].
Hence, once we obtained the selenoprotein gene in Vipera berus, we used SecisSearch3 software to predict SECIs elements in the genomic region of each protein, giving as inout the genomic.fa file and using the three available methods (Infernal, Covels and Original) to perform an optimized and restrictive SECIs search.
Finally, we extracted the absolute coordenates of the SECIs elements from their relative positions of the $sample.genomic.fa with a script for the selenoproteins and another one for the homologous in cys.
As we obtained multiple SECIs elements for gene, we selected the best one according the following criteria:
1. SECIs element and gene must come from the same frame.
2. SECIs elements have to be located 3' nearby the last exon.
![]()
© Universitat Pompeu Fabra
Plaça de la Mercè, 10-12. 08002 Barcelona
☎ (+34) 93 542 20 00
{kind=link}