
Per obtenir les nostres seqüències query, vam generar un arbre filogenètic de les espècies contingudes en la base de dades SelenoDB, amb l'objectiu de trobar l'espècie més propera a Vipera berus que tingui els gens codificants per selenoproteïnes ja predits. L'arbre es va generar amb el programa PhyloT, el qual genera arbres filogenètics basats en la taxonomia de NCBI (disponible online: http://phylot.biobyte.de/) i, mitjançant el software Interactive Tree Of Life (iTOL), es va visualitzar[12][13].
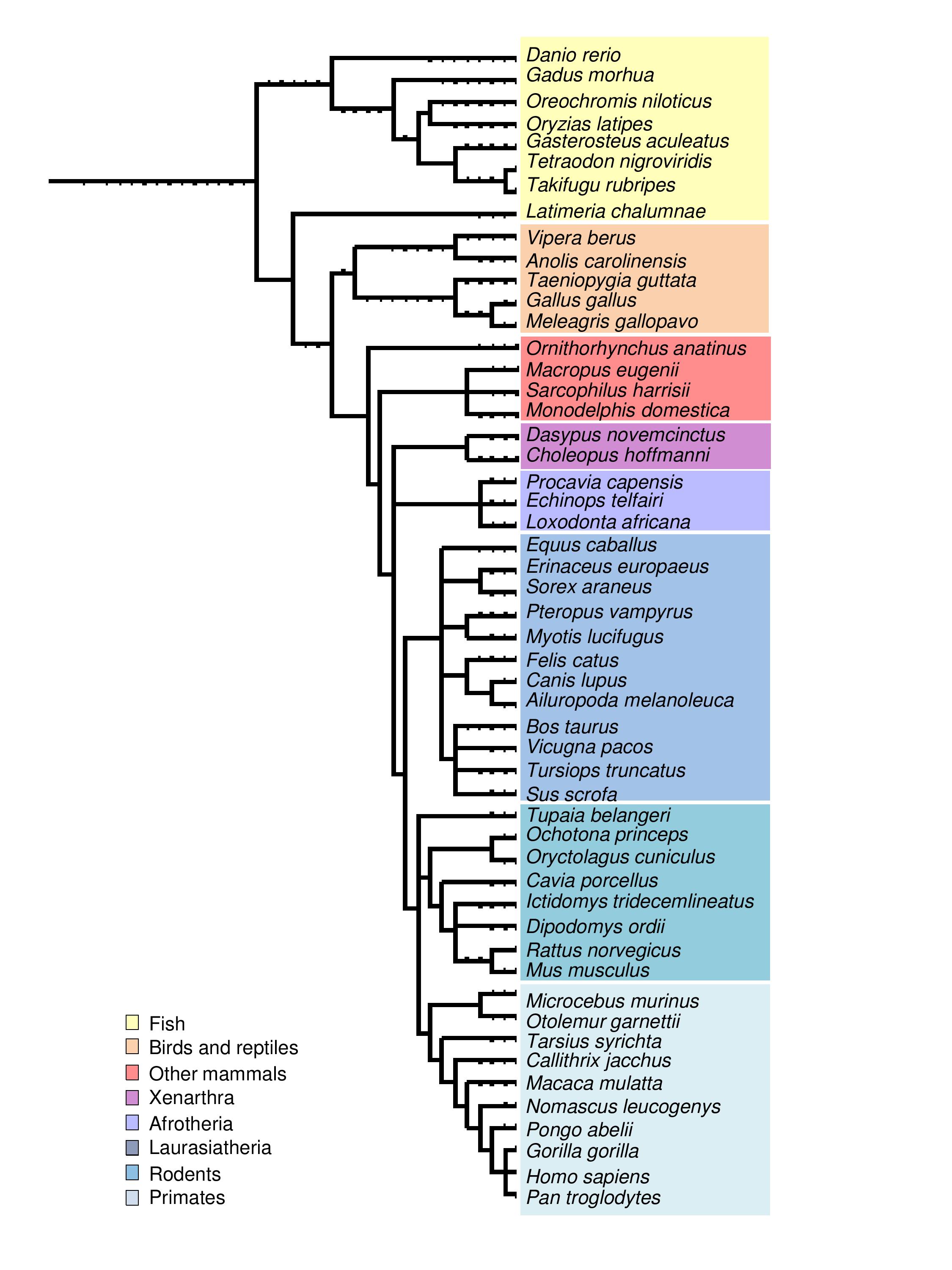
Figura 1. Arbre filogenètic de les espècies de SelenoDB i Vipera berus.
Com es pot veure en la Figura 1, l'espècie més propera a Vipera berus en la base de dades SelenoDB és Anolis carolinensis i, per tant, les seqüències predites per les seves selenoproteïnes ens van servir com a querys. Aquestes seqüències les vam dividir en red proteins (si contenien una selenocisteïna en la seva seqüència) i green proteins (si eren homòlogues que contenien cisteïna o proteïnes que participen en la via de síntesi de selenoproteïnes).
Clica aquí per entendre el sistema de fitxers utilitzats en aquest treball, on els rectangles blaus són carpetes i els blancs fitxers de text. Això és d'especial importància per entendre els paths utilitzats en els scripts.
Després d'obtenir les querys d'Anolis carolinensis, vam canviar automàticament les U per X en la seqüència de les red_proteins, per evitar errors posteriorment (script en bash).
Un cop vam tenir les nostres querys, vam córrer un tblastn contra el genoma de Vipera berus. La modalitat tblastn agafa una seqüència d'aminoàcids com a query i la compara amb el genoma de referència traduït en els seus 6 possibles marcs de lectura. Vam automatitzar el procés escrivint un script en llenguatge BASH, que ens va permetre obtenir l'output de cada blast realitzat per cada proteïna. Els scripts es poden trobar en els següents enllaços: red_proteins i green_proteins.
Un cop obtinguts els outputs del BLAST, vam prosseguir a analitzar-los un per un manualment. La selecció manual dels hits ens va permetre tenir en compte diferents paràmetres i no només posar un valor llindar d'E-value de manera automàtica. Així doncs, per seleccionar els millors hits vam seguir els següents passos:
1. Selecció dels millors hits i descart dels pitjors. Ens vam fixar en l'E-value, el qual indica quantes vegades s'espera trobar un hit amb el mateix score o major per atzar: com més baix sigui, per tant, més improbable serà que el resultat sigui conseqüència de l'atzar. Si un E-value era menor a 0,001, el teníem en consideració i seguíem analitzant altres paràmetres.
2. Observació i anàlisi dels alineaments. Teníem en compte el següent:
2.1. E-value: dins de cada scaffold, hi pot haver diversos hits, cadascun amb el seu E-value. Si algun d'aquests valors era elevat el descartàvem.
2.2. Alineament de la selenocisteïna (U): volem que la U de la nostra query s'alineï amb un codó STOP (símbol *) o bé amb una Cys.
2.3. Solapament o no dels diferents alineaments: la nostra hipòtesi és que els hits que es troben a la mateixa seqüència corresponen a diferents exons del mateix gen i, per tant, s'espera que l'últim aminoàcid alineat d'un hit coincideixi amb el primer del següent hit, amb el mínim de solapaments. Tenim en compte, també, que la distància al genoma de referència entre els diferents alineaments s'ha de correspondre amb la llargada d'un intró de Vipera berus. Com que el genoma de Vipera berus encara no està anotat, hem utilitzat la llargada mitja d'un intró d'un transcrit codificant d'Anolis carolinensis, la qual és de 5041 bp (min: 9bp; màx: 856457 bp)[14].
2.4. Marc de lectura: en unir els hits per un mateix scaffold, també teníem en compte que es trobessin en el mateix marc de lectura (positiu o negatiu). A més, les posicions d'inici i final depenen de si aquest és positiu o negatiu.
3. Quan un hit complia els nostres criteris, guardàvem en un nou fitxer l'identificador del contig, l'inici i el final de la seqüència. Aquest fitxer seria usat posteriorment per predir el nostre gen.
La resta del procés per a la cerca de Selenoproteïnes en Vipera berus s'ha realitzat de manera automàtica mitjançant un script en bash per a les selenoproteïnes (red_proteins) i per les homòlogues en Cys/maquinària (green_proteins).
Un cop s'han seleccionat els hits, cal extreure la regió d'interès a partir del genoma de Vipera berus. Primer, cal obtenir la seqüència que conté la regió mitjançant el fastafetch:
Després, s'ha d'extreure la regió a partir de la seqüència (output del fastafetch), ja que fastasubseq només funciona sobre un fitxer FASTA que contingui una única seqüència:
Estimació de start i length: a partir del valor d'inici i final del hit, s'estenen 50,000 nucleòtids per banda, per tal d'extreure la regió d'interès que contingui tot el gen. Sempre tenint en compte que el valor calculat d'inici no sigui menor de 1 i el de length major que la length del contig (fitxer lengths.fa).
A partir de la regió d'interès de cada hit, vam procedir a predir l'estructura del gen:
- m p2g: mode de proteïna a gen
- showtargetgff: format GFF en l'output
- q: query (proteïna d'Anolis carolinensis)
- t: target (regió d'interès de Vipera berus)
A més, sobre la predicció de tot el gen, vam extreure només aquelles regions relatives als exons:
Un cop obtingut el fitxer en format gff, cal convertir-lo en format fasta mitjançant el programa en perl fastaseqfromGFF.pl:
Per últim, vam traduir la seqüència predita de cDNA a proteïna:
La opció -F indica forward i la posició del nucleòtid on s'iniciarà la traducció. La predicció de la proteïna es fa a partir de cDNA (exons) i, per tant, es pot fer servir la opció -F 1.
El fastatraslate, quan troba un codó stop TGA, col•loca un *. Això pot suposar que el tcoffee generi un mal alineament i, per tant, cal canviar els * per X. D'aquesta manera, en el tcoffee podrem veure la selenocisteïna de la query (també canviada per X) amb un TGA o bé amb una cisteïna de la proteïna predita.
Després d'obtenir la predicció de la seqüència d'aminoàcids de la proteïna, la vam comparar amb la query de la mateixa proteïna d'Anolis carolinensis, mitjançant un alineament generat pel programa t-coffee.
Pel que fa a les proteïnes homòlogues en Cys, cal fer un alineament previ d'aquestes amb les selenoproteïnes homòlogues, per identificar en quina posició es troba la C i, així, determinar si s'alinea amb la nostra predicció en l'output de la comparació entre la proteïna predita i la query.
Per tal d'obtenir les coordenades absolutes respecte el contig dels gens i exons predits per l'exonerate, vam crear un script que automatitzés el càlcul: es té en compte l'inici donat en el fastasubseq i s'hi suma la posició relativa obtinguda en l'output de l'exonerate ($sample.exonerate.gff). Així, s'aconsegueixen les posicions absolutes tant dels gens com de cadascun dels exons predits. En els següents enllaços es pot accedir als scripts: coordenades_gens_absolutes.sh i coordenades_exons_absolutes.sh per les red_proteins, coordenades_gensgreen_absolutes.sh i coordenades_exonsgreen_absolutes.sh per les green_proteins.
Per últim, vam anotar també en quina posició d'aminoàcid de la proteïna predita es troba la selenocisteïna o la cisteïna alineades.
SecisSearch3 és un mètode computacional que reemplaça el seu predecessor SecisSearch per a la predicció d'elements SECIs eucariòtics. És un software públic i disponible a: http://gladyshevlab.org/SelenoproteinPredictionServer/[15].
Així doncs, després d'obtenir el gen de les selenoproteïnes en Vipera berus, hem utilitzat el software SecisSearch3 per predir elements SECIs presents en la regió genòmica de cada proteïna, donant com a input el genomic.fa i utilitzant els tres mètodes disponibles (Infernal, Covels I Original), per tal de fer una cerca de SECIs més restrictiva i optimitzada.
Finalment, un cop obtinguts els elements SECIs, hem extret les seves coordenades absolutes a partir de les posicions relatives del $sample.genomic.fa, mitjançant un script per les selenoproteïnes i un altre per les homòlogues en cys.
Vam obtenir múltiples elements SECIs en SeciSearch3 i, per tant, en vam seleccionar un per gen tenint en compte els següents criteris:
1. Els SECIs i el gen han d'estar en el mateix marc de lectura.
2. Els SECIs s'han de situar 3' propers a l'últim exó.
![]()
© Universitat Pompeu Fabra
Plaça de la Mercè, 10-12. 08002 Barcelona
☎ (+34) 93 542 20 00
{kind=link}