
Para obtener nuestras secuencias query, generamos un árbol filogenético de las especies anotadas en la base de datos SelenoDB, con el objetivo de encontrar aquella especie más cercana a Vipera berus que presente los genes codificantes para selenoproteínas ya predichos. El árbol se creó con el programa PhyloT, que genera árboles filogenéticos basados en la taxonomía de NCBI (disponible online: http://phylot.biobyte.de/) y, mediante el software Interactive Tree Of Life (iTOL), se visualizó[12][13].
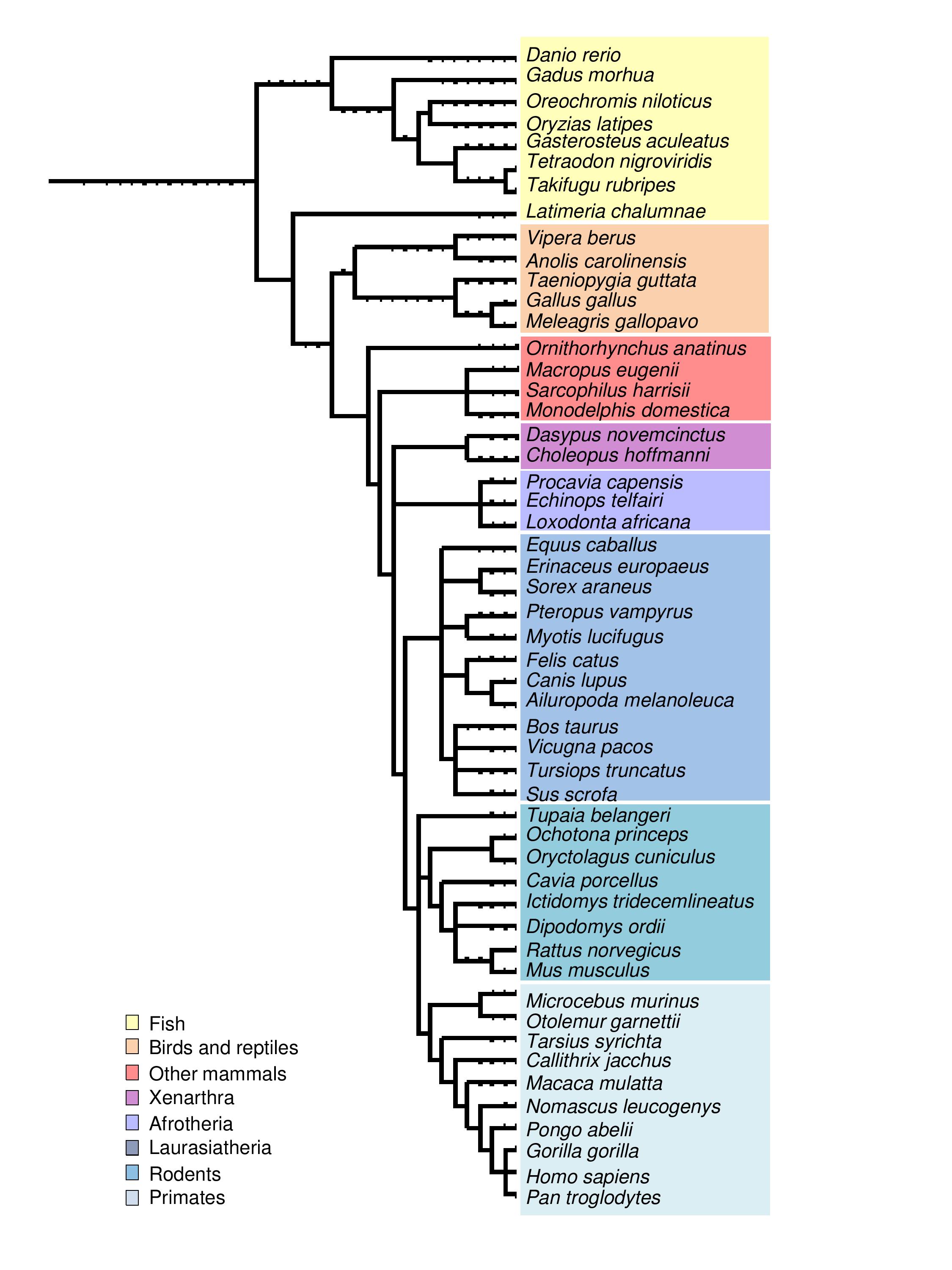
Figura 1. Árbol filogenético de las especies de SelenoDB y Vipera berus.
Como muestra la Figura 1, la especie más cercana a Vipera berus en la base de datos SelenoDB es Anolis carolinensis y, en consecuencia, utilizamos las secuencias predichas de sus selenoproteínas como querys. Dividimos estas secuencias en red proteins (si contenían una selenocisteína) y green proteins (si eran homólogas con cisteína o proteínas que participan en la vía de síntesis de selenoproteínas).
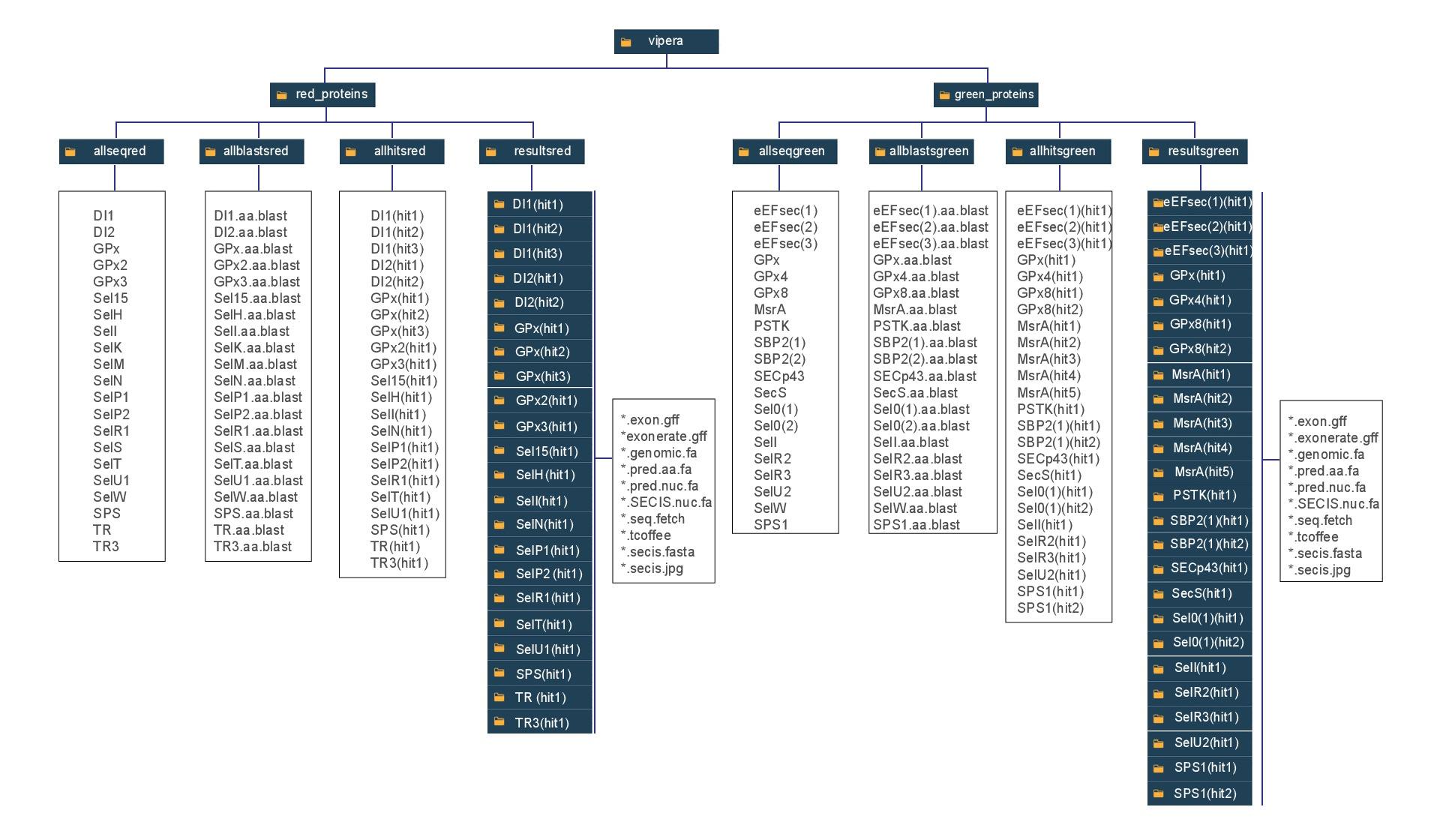
Clica aquí para entender el sistema de ficheros utilizado en este trabajo, dònde los rectángulos azules son carpetas y los blancos, ficheros de texto. Esto es de especial importancia para entender los paths utilizados en los scripts.
Después de obtener las querys de Anolis carolinensis, cambiamos automáticamente las U por X en la secuencia de las red_proteins, para evitar errores posteriormente (script en bash).
Una vez obtenidas las querys, realizamos un tblastn contra el genoma de Vipera berus. La modalidad tblastn utiliza una secuencia de aminoácidos de query y la compara con el genoma de referencia traducido en sus 6 posibles marcos de lectura. Automatizamos el proceso con un script en lenguaje BASH, que nos permitió obtener el output de cada blast realizado para cada proteína. Los scripts se pueden encontrar en los siguentes enlaces: red_proteins y green_proteins.
Una vez obtenidos los outputs del BLAST, se prosiguió a analizarlos uno por uno manualmente. La selección manual de los hits nos permitió tener en cuenta diferentes parámetros y no sólo poner un valor umbral de E-value umbral automáticamente). Así, para seleccionar los mejores hits seguimos los siguientes pasos:
1. Selección de los mejores hits y descarte de los peores. Nos fijamos en el E-value, que indica cuántas veces se espera encontrar un hit con el mismo score o mayor por azar: como más bajo sea, pot tanto, más improbable será que el resultado sea consecuencia del azar. Si un E-value era menor de 0,001, lo seleccionábamos y segu�amos analizando los otros parámetros.
2. Observación y análisis de los alineamientos. Teníamos en cuenta:
2.1. E-value: dentro de cada scaffold, puede haber múltiples hits, cada uno con su E-value. Si alguno era elevado lo descartábamos.
2.2. Alineamiento de la selenocisteína (U): queremos que la U de nuestra query se alinee con un codón STOP (símbolo *) o bien con una Cys.
2.3. Solapamiento o no de los diferentes alineamientos: nuestra hipótesis es que los hits que se encuentran en la misma secuencia corresponden a diferentes exones del mismo gen y, por tanto, se espera que el último aminoácido alineado de un hit coincida con el primero del siguiente hit, con el mínimo de solapamientos. También teníamos en cuenta que la distancia al genoma de referencia entre los diferentes alinemientos tiene que corresponderse con la longitud de un intrón de Vipera berus. El genoma de Vipera berus no está anotado aún y, por este motivo, hemos utilizado la longitud media de un intrón de un transcrito codificante de Anolis carolinensis, la cual es de 5041 bp (min: 9bp; máx: 856457 bp)[14].
2.4. Marco de lectura: al unir los hits de un mismo scaffold, también tuvimos en cuenta que se encontrasen en el mismo marco de lectura (positivo o negativo). Además, las posiciones de incio y final dependen de si éste es positvo o negativo.
3. Cuando un hit cumplía los criterios descritos, guardábamos en un nuevo fichero el identificador del contig, el inicio y el final de la secuencia. Este fichero sería utilizado posteriormente para predecir nuestro gen.
El resto del proceso para la búsqueda de Selenoproteínas en Vipera berus se ha realizado de manera automática mediante un script en bash para las selenoproteínas (red_proteins) y para las homólogas en Cys/maquinaria (green_proteins).
Una vez seleccionados los hits, se tiene que extraer la región de interés a partir del genoma de Vipera berus. Primero, se debe obtener la secuencia que contiene la región:
Después, se extrae la región a partir de la secuencia (output del fastafetch), ya que fastasubseq sólo funciona sobre un fichero FASTA que contenga una única secuencia:
Estimación de start y length: a partir del valor de inicio y final del hit, se extienden 50,000 nucleótidos por banda, para extraer la región de interés que contenga todo el gen. Siempre teniendo en cuenta que el valor calculado de inicio no sea menor de 1 y el de length mayor que la length del contig (fichero lengths.fa).
A partir de la región de interés de cada hit, procedimos a predecir la estructura del gen:
- m p2g: modo de proteína a gen
- showtargetgff: formato GFF en el output
- q: query (proteína de Anolis carolinensis)
- t: target (región de interés de Vipera berus)
Además, sobre la predicción de todo el gen, extrajimos sólo aquellas regiones relativas a los exones:
Una vez obtenido el fichero en formato gff, hay que convertirlo en formato fasta mediante el programa en perl fastaseqfromGFF.pl:
Por último, tradujimos la secuencia predicha de cDNA a proteína:
La opción -F indica forward i la posición del nucleótido donde se iniciará la traducción. La predicción de la proteína se hace a partir de cDNA (exones) y, por tanto, se puede utilizar la opción -F 1.
El fastatraslate, cuando encuentra un codón stop TGA, coloca un *. Esto puede suponer que el tcoffee genere un mal alineamiento y, por tanto, se tienen que cambiar los * por X. De esta manera, en el tcoffee podremos ver la selenocisteína de la query (también cambiada por X) con un TGA o bién con una cisteína de la proteína predicha.
Después de obtener la predicción de la secuencia de aminoácidos de la proteína, la comparamos con la query de la misma proteína de Anolis carolinensis, mediante un alineamiento generado por el programa t-coffee.
Respecto a las proteïnas homólogas en Cys, se tiene que hacer un alineamiento previo de estas con las selenoproteínas homólogas, para identificar en qué posición se encuentra la C y, así, determinar si se alinea con nuestra predicción en el output de la comparación entre la proteína predicha y la query.
Para obtener las coordenadas absolutas respecto del contig de los genes y exones predichos por el exonerate, creamos un script que automatizara el cálculo: se tiene en cuenta el inicio dado en el fastasubseq y se le suma la posición relativa obtenida en el output del exonerate ($sample.exonerate.gff). Así, se consiguen las posiciones absolutas tanto de los genes como de cada uno de los exones predichos. En los siguientes enlaces se puede acceder a los scripts: coordenades_gens_absolutes.sh y coordenades_exons_absolutes.sh para las red_proteins, coordenades_gensgreen_absolutes.sh y coordenades_exonsgreen_absolutes.sh para las green_proteins.
Por último, anotamos también en qué posición de aminoácido de la proteína predicha se encuentratroba la selenocisteína o la cisteína alineadas.
SecisSearch3 es un método computacional que reemplaza su predecesor SecisSearch para la predicción de elementos SECIs eucarióticos. Es un software público y disponible en: http://gladyshevlab.org/SelenoproteinPredictionServer/ [15].
Así pues, después de obtener el gen de las selenoproteínas en Vipera berus, hemos utilizado el software SecisSearch3 para predecir elementos SECIs presentes en la región genómica de cada proteína, dando como input el genomic.fa y utilizando los tres métodos disponibles (Infernal, Covels I Original), para hacer una búsqueda de SECIs más restrictiva y optimizada.
Finalmente, una vez obtenidos los elementos SECIs, hemos extraído sus coordenadas absolutas a partir de las posiciones relativas del $sample.genomic.fa, mediante un script para las selenoproteínas y otro para las homólogas en cys.
Obtubimos múltiples elementos SECIs en SeciSearch3 y, por tanto, seleccionar uno por gen teniendo en cuenta los siguientes criterios:
1. Los SECIs y el gen tienen que estar en el mismo marco de lectura.
2. Los SECIs se tienen que situar 3' próximos al último exón.
![]()
© Universitat Pompeu Fabra
Plaça de la Mercè, 10-12. 08002 Barcelona
☎ (+34) 93 542 20 00
{kind=link}