Materials i mètodes
L'objectiu del nostre treball és identificar les selenoproteïnes EhSEP2, Sel O i Sel S en el genoma de diferents protistes. El protocol del treball s'ha estructurat en diferents apartats que es corresponen amb els diferents programes que hem utilitzat.
- Obtenció de les querys.
- Obtenció dels genomes.
- Localització d’una regió del genoma: tBlastn.
- Extracció de la regió genòmica on potencialment es troba el nostra gen.
- Generació de l’anotació d’un gen: obtenció dels exons.
- Obtenció del cDNA i de la seqüència proteica.
- Alineament global.
- Realització d'un Blastp
- Cerca d'elements SECIS.
- Maquinària.
1. Obtenció de les querys
El nostre objectiu per a l’obtenció de les querys, va ser trobar aquelles que fossin més properes als nostres protistes.
La query de EhSEP2 (E.Huxley), va ser aportada pel professorat de l’assignatura. Per obtenir la de Sel O, primer vam utilitzar la proteïna homòloga a Sel O de P.infestans, obtinguda en cursos anteriors, però vam decidir fer un BlastP amb aquesta seqüència en la web del NCBI per intentar tenir-ne una de més propera. En fer-ho ens vam adonar que hi havia la seqüència homòloga a Sel O en E.siliculosus, que de fet es tracta d’uns dels organismes que havíem d’analitzar. A més a més, hem utilitzat també una query procariota, concretament la del microorganisme A.anophagefferens. Així doncs, la complexitat de la familia Sel O ens ha obligat a utilitzar aquestes tres querys per poder obtenir els millors resultats possibles.
Per a la obtenció de la query de Sel S vam anar a la pàgina de PhylomeDB. Vam buscar Sel S i vam obtenir un arbre filogenètic on vam veure que l'organisme més proper als nostres protistes era C.intestinalis, un urocordat filogenèticament allunyat de la resta d'organismes que contenen Sel S.
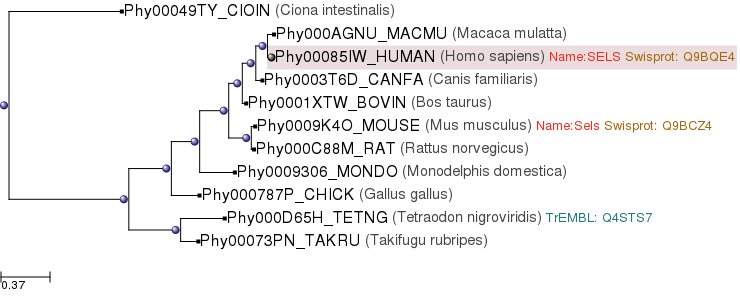{kind=link}
En tots els casos vam comparar els blasts obtinguts amb aquestes querys, amb la query d’H.sapiens i S.cerevisiae però finalment ens vam decantar per les querys explicades anteriorment.
2. Obtenció dels genomes
Els genomes de tots els microorganimes van ser proporcionats pel professorat i es troben en els ordinadors de la Universitat en la següent direcció:
/cursos/BI/genomes/protists/2011/N.organisme/genome.fa
3. Localització d’una regió del genoma: tBLASTn
El BLAST (Basic Local Alignment Search Tool) és un algorisme heurístic que compara seqüències biològiques per tal de fer alineaments locals.
Per tal de localitzar la nostra seqüència query en els genomes dels organismes, hem utilitzat el tBLASTn. Aquest tipus de BLAST s’encarrega de comparar la nostra query proteica en una base de dades genòmica, transformant-la i fent l’alineament en seqüències d’aminoàcids. Aquest software el trobem a les aules d’informàtica de la universitat.
Primer de tot, hem de introduir les següents ordres al terminal per tal d’executar el programa:
$ export PATH=$PATH:/disc8/bin/ncbiblast/bi
$ cp /cursos/BI/bin/ncbiblast/.ncbirc ~/
A continuació utilitzem la següent comanda:
$blastall -p tblastn -i query_nomselenoproteina.fa -d /cursos/BI/genomes/protists/2010/N.organisme/genome.fa -o tblastnA_selenoproteinaANDN.organisme.fa -F F -e 0.00001
En aquesta comanda observem els següents arguments:
- -p: indica el tipus de BLAST que estem utilitzant.
- -i: serveix per especificar la nostra query.
- -d: serveix per especificar l’arxiu on es troba el genoma de l'organisme on volem fer la cerca. Nosaltres utilitzem com a base de dades l’arxiu que conté tot el genoma del protist, contra el qual compararem la nostra query.
- -o: serveix per anomenar l’arxiu de sortida.
- -F F: elimina les regions de baixa complexitat, fet que augmenta la qualitat de l’alineament.
- -e: fixa el valor màxim del E-value, de manera que tots els valors més alts seran obviats. En el nostre cas, hem fixat l’E-value en 0,00001. Cal dir que, depenent de la familia de selenoproteïnes, aquest valor s’ha suavitzat una mica, ja que sinó no obteníem cap resultat. Això es comentarà amb més precisió en la familia corresponent.
En aquest fitxer podem observar els alineaments i els E-value, de manera que podrem fer-nos una idea d'allò que tenim, abans de continuar. Per fer els següents passos necessitarem paràmetres que en aquest fitxer no apareixen, per tant, creem un nou fitxer amb un format una mica diferent, utilitzant -m 9 a més dels anteriors arguments. En els fitxers obtinguts apareixeran les dades en forma de taula. La comanda utilitzada és la següent:
$ blastall -p tblastn -i query_nomselenoproteina.fa -d /cursos/BI/genomes/protists/2011/N.organisme/genome.fa -o tblastnA_selenoproteinaANDN.organisme.fa -m 9 -F F -e 0.00001
En aquest apartat cal comentar que, per tal de realitzar el BLAST, la base de dades ha d’estar formatejada, en el nostre cas els genomes. Per això cal introduir prèviament la següent comanda:
$ formatdb -i /cursos/BI/genomes/protists/2011/N.organisme/genome.fa -p F -n N.organime.fa
El procés d’obtenció d’aquests fitxers ha estat automatitzat mitjançant dos programes: el tBLASTnA i el tBLASTnB.
En aquest cas, l'argument -i indica on es troba el genoma de l'organisme; el paràmetre -p F informa que la base de dades que fem servir no és un arxiu proteic, sinó que conté DNA. Finalment, l'argument -n ens permet renombrar l'arxiu de sortida de la base de dades.
Aquest procés no l’hem realitzat, ja que els genomes dels organismes ja estaven formatejats.
4. Extracció de la regió genòmica on potencialment es troba el nostra gen
Un cop escollit l'alineament o alineaments que són d'interès es procedeix a l'extracció de la seqüència genòmica del contig significatiu. Per fer-ho necessitem una sèrie de programes que formen part d’un software anomenat exonerate.
Per tal de poder executar tots els programes que pertanyen a l’exonerate, cal introduir en el terminal la següent comanda:
$ export PATH=/cursos/BI/bin/exonerate/i386/bin:$PATH
El nostre objectiu és extreure d’un fitxer multifasta, un únic fitxer fasta que contingui la regió del genoma on el BLAST ha predit que es troba el nostre gen d’interès.
Els programes que han estat:
- Fastaindex (acompanya a l’Exonerate)
El primer que cal fer és indexar (ordenar i enumerar) el genoma del protist que s'utilitza com a base de dades mitjançant la comanda següent:
$fastaindex /cursos/BI/ genomes/protists/any/N.protista/genome.fa N.organisme.index
En el nostre cas, aquest fitxer ens el va proporcionar la universitat a la següent direcció:
/cursos/BI/genomes/protists/any/N.organisme/genome.index
- Fastafetch (acompanya a l’Exonerate)
A continuació, es procedeix a extreure la regió genòmica d'interès. Es comença per delimitar el scaffold on tenim la proteïna per mitjà del programa fastafetch amb la comanda que s’expressa a continuació:
$fastafetch/cursos/BI/genomes/protists/any/N.protista/genome.fa cursos/BI/genomes/protists/any/nom_protista/genome.index 'identificador del contig' > fastafetchselenoproteinaANDN.organisme.fa
- Fastasubsequence (acompanya a l’Exonerate)
Un cop ja hem delimitat el scaffold on tenim la proteïna, dins d'aquest cal acotar millor la regió on es troba el gen d'interès. Per això, amb una seqüència més curta podrem treballar amb més facilitat. Per assegurar-nos que agafem el gen sencer i que s'inclouen els extrems 3' i 5' agafarem una regió upstream i downstream d'entre 25 i 25 kbp. En alguns casos hem variat aquesta longitut.
Per fer-ho, vam utilitzar la següent comanda:
$fastasubseq fastafetchqueryANDNOM.organisme.fa inici longitud > fastasubseq_selenoproteinaANDN.organisme.fa
Com a nucleòtid d'inici vam agafar el més petit, independentment de si l'alineament es produïa en sentit forward o reverse.
5. Generació de l’anotació d’un gen: obtenció dels exons
Per obtenir l’anotació del gen utilitzarem un seguit de programes que també formen part de l'exonerate.
A continuació, amb la seqüència extreta dels genomes, on potencialment es troba el nostre gen d’interés, generarem l’anotació exònica que donarà lloc a la proteïna. Això ho fem cridant el programa exonerate de la següent manera:
$ exonerate -m p2g --showtargetgff --exhaustive yes -q query_nomselenoproteina.fa -t fastasubseq_selenoprotreinaANDN.organisme.fa
En aquesta comanda observem els següents arguments:
- -m: especifica el tipus d'alineament. En el nostre cas és p2g que vol dir protein to genome.
- --showtargetgff: extreu els resultats en format .gff
- --exhaustive yes: obté resultats més acurats.
- -q: especifica la seqüències de query.
- -t: especifica la seqüència a comparar.
A l’hora d’executar aquest programa, cal tenir en compte que la query utilitzada no pot contenir el símbol U (selenocisteïna), ja que el programa no el reconeix. Per aquest motiu, l’hem canviat per una X en els casos en que la nostra query contenia una selenocisteïna. Quan la query utilitzada és una proteïna homòloga, no cal aquest canvi, ja que la C sí que la reconeix.
Ens interesa extreure la seqüència de cDNA, per tant, només necessitem els exons i per això utilitzem la comanda egrep -w exon. L'ordre utilitzada ha estat la següent:
$ exonerate -m p2g --showtargetgff --exhaustive yes -q query_nomselenoproteina.fa -t fastasubseq_queryselenoprotreinaANDN.organisme.fa | egrep -w exon > alineament_selenoproteina_N.organisme.exonerate.gff
D’aquesta manera conseguirem extreure tots els exons i posar-los en un arxiu en format .gff, que serà utilitzat per poder obtenir el cDNA.
Finalment, comentar que també hem utilitzat un altra programa per a la generació de l’anotació genòmica. En els casos en que l’exonerate no ens ha funcionat correctament hem utilitzat el Genewise. Les comandes utilitzades han estat:
$ export PATH=/disc8/bin:$PATH
$ export WISECONFIGDIR=/disc8/soft/wise-2.2.0/wisecfg
Aquestes primeres comandes serveixen per a executar el programa. A continuació, quan teniem una cadena forward hem utilitzat:
$ genewise -pep -pretty -cdna -gff query_selenoproteina.fa fastasubseq_selenoproteinaAND_N.organisme.fa > genewise_selenopreteinaANDN.organisme.fa
En els casos que teniem una cadena reverse hem utilitzat la següent comanda:
$ genewise -pep -pretty -cdna -gff -trev query_selenoproteina.fa fastasubseq_selenoproteinaANDN.organisme.fa > genewise_selenoproteinaANDN.organisme.fa
El pas corresponent al exonerate l'hem automatitzat mitjançant un programa informàtic.
6. Obtenció del cDNA i de la seqüència proteica
El programa utilitzat per fer aquest pas és el fastaseqfromGFF.pl que està a la nostra disposició a les aules d’informàtica. Primerament, haurem d’executar el programa:
export PATH=/cursos/BI/bin:$PATHH
A continuació, la ordre que em de donar-li al terminal és:
$ fastaseqfromGFF.pl fastasubseq_selenoprotreinaANDN.organisme.fa alineament_nomselenoproteina_N.organisme.exonerate.gff > cDNA_N.organisme.fa
Aquest pas l'hem automatitzat amb el següent programa.
L’arxiu obtingut, anomenat cDNA_N.organisme.fa, com el seu nom ens indica, conté el cDNA. A nosaltres el que ens interessa obtenir és la seqüència peptídica de la nostra hipotètica proteïna. Per aquest motiu hem utilitzat un altra programa que incorpora el software de l’exonerate. És el fastatranslate, que ens permet obtenir la proteïna amb totes les pautes de lectura possibles. La comanda introduïda al terminal és la següent:
$ fastatranslate cDNA_N.organisme.fa > proteina_selenoproteinaAND_N.organisme.fa
Per tal de que el programa ens mostrés només la millor proteïna obtenida hem afegit la comanda -F 1:
$ fastatranslate - F 1 cDNA_N.organisme.fa > proteina_selenoproteinaAND_N.organisme.fa
Aquest procés també l’hem automatitzat amb un programa.
7. Alineament global
Gràcies a la utilització del programa TCOFFEE hem pogut realitzar un alineament global de la proteïna obtinguda amb la nostra query. Això ens ha permès fer-nos una idea de que era la proteïna que havíem obtingut.
Per fer aquest alineament hem introduit, tant la query com la proteïna obtinguda, dins d’un fitxer. A continuació utilitzem la comanda:
$ t_coffee nomfitxer.fa > tcoffee_selenoproteina_N.organisme.fa
Però amb aquest alineament no en tenim prou per extreure conclusions definitives. Per aquest motiu, un cop obtingudes totes les sequències proteiques de cada query, hem fet un alineament global de totes elles amb la query. El procediment ha estat el mateix que el realitzat anteriorment, però en aquest cas el fitxer contenia totes les seqüències proteiques, a més de la query. La comanda utilitzada ha estat:
t_coffee nomfitxer.fa > alineamentglobal_nomselenoproteina.fa
8. Realització d'un BLASTp
Per tal d’assegurar-nos que la proteïna que hem obtingut forma part de la familia que cercàvem hem realitzat un BLASTp a la pàgina web de NCBI. D’aquesta manera hem pogut saber si la proteïna obtinguda era la que nosaltres buscàvem o no.
Dels resultats obtinguts ens hem fixat ens el motius conservats i en el primers hits. És a dir, que la proteïna obtinguda, tingués els mateixos motius conservats que la nostra query, i a més a més, els millors hits fossin proteïnes de la mateixa familia.
9. Cerca d'elements SECIS
Un cop obtinguts els resultats, per confirmar la presència de selenocisteïnes, o l'absència d'aquestes, hem decidit cercar els elements SECIS. La cerca l'hem realitzat tant en presència de selenoproteïnes o homòlogues, així com en la seva absència. En aquells casos on no podíem continuar el procés i obtenir una proteïna, s’han cercat SECIS, per tal de demostrar que no hi havia cap selenoproteïna en aquella regió.
Hi ha dues maneres que ens permeten cercar SECIS:
La primera, i la que hem utilitzat nosaltres, és la introducció del fitxer obtingut en el fastasubseq a la pàgina web SECISearch, mirant també si en trobàvem algun a la cadena complementària. Els modes utilitzats han estat l’Strict, i en alguns casos, es va optar per rebaixar els nivells d’energia requerits per obtenir SECIS (Loose canonical). Aquesta pàgina web, si hi ha elements SECIS ens mostra la seqüència que els defineix, una imatge de l’estructura d’aquest element i també l’score. Nosaltres vam optar per obsevar els scores obtinguts, i escollir el més alt, en cas que fos prou significatiu.
L’altra opció per tal de cercar els elements SECIS, és la utilització d’un programa que es pot córrer des del terminal de l’ordinador mitjançant les comandes següents.
Aquesta comanda serveix per cridar el programa:
$ export PATH=/cursos/BI/bin:$PATH
Amb aquesta comanda obtenim els arxius:
$ SECISearch.pl fastasubseq_querynomselenoprotreinaANDN.organisme.fa > secisnomselenoproteinaANDN.organisme.fa
El tutors ens van aconsellar que féssim servir la pàgina web, i per aquest motiu nosaltres hem utilitzat el primer mètode.
10. Maquinària
Un cop havíem finalitzat l’anàlisi de les tres famílies de selenoproteïnes, ens vam plantejar si aquests organismes protists disposaven de maquinària per a la síntesi d’aquestes. Cal dir, que la presència d’aquesta maquinària, no invalida els resultats obtinguts en cas de no haver obtingut cap selenoproteïna, ja que es tracta de proteïnes que es requereixen per la síntesi de qualsevol selenoproteïna. Conseqüentment, els organismes que tinguin alguna selenoproteïna disposaran de maquinària, encara que les selenoproteïnes no siguin de les nostres famílies.
Les proteïnes que es coneix que serveixen per a la síntesi són:
- SPS1 (Selenofosfat Sintasa 1)
- SPS2 (Selenofosfat Sintasa 2)
- SecS (SLA/LP)
- Sec43p
- SBP2
- eEFsec
- PSTK
Per cercar totes aquestes proteïnes en els genomes dels organismes s’ha realitzat el mateix procés que en la cerca de les famílies de selenoproteïnes, utilitzant com a querys, les seqüències proteiques d’altres organismes obtingudes a SelenoDB i NCBI.
A partir d’aquí, el procés seguit ha estat el mateix, amb cadascun dels organismes i amb cadascuna de les proteïnes (punts 3,4,5,6). A continuació, de la mateixa manera que vam fer amb les famílies de selenoproteïnes assignades, vam realitzar una alineament global, per veure la conservació de les diferents seqüències.
Finalment, vam realitzar un BLASTp, per veure si la proteïna obtinguda era realment el que buscàvem. En SPS1/2 vam buscar els elements SECIS.
En un principi vam intentar cercar tRNAs a partir de la pàgina web tRNAscan-SE Search Server però no ens en vam ensortir. Finalment, i després de varis intents nuls, el professorat ens va aportar els resultats dels tRNAs corresponents als organismes estudiats al llarg de tot el projecte en qüestió, correguts amb el tRNAscan. L’objectiu d’aquest programa és escannejar tots els tRNAs presents en el genoma de cada organisme.
Els resultats obtinguts dels tRNAs es mostren en la pàgina de resultats i discussió a dins de l'apartat de maquinària que mostra els organismes ordenats alfabèticament. Per tal de cercar els tRNAs, buscarem el codó complementari i revers a l’anticodó. És a dir, el codó corresponent a la selenocisteïna és el TGA, per tant, l’anticodó que codifica per la selenocisteïna és el TCA.
Finalment, cal dir que aquests procediments han patit certes modificacions depenent de la família de selenoproteïnes estudiada i dels resultats que anàvem obtenint. Els detalls més exhaustius seran explicats en l’apartat de discussió i resultats en cada cas.