ANÀLISI DE SEQÜÈNCIES: regió CFTR
Laia Morató i Clara Panosa
Facultat de Ciències de la Vida i la Salut, UPF
ABSTRACT
El treball que presentem a continuació pretén analitzar una regió del genoma humà utilitzant una aproximació del protocol que segueix el projecte Encode.
La seqüència que analitzem és la de la regió que comprèn el gen CFTR que té una allargada aproximada de 2Mb i es troba al cromosoma 7. Utilitzant, doncs, programes informàtics de predicció de gens intentem caracteritzar la seqüència trobant regions que codifiquen per gens i els seus exons. Validem els resultats obtinguts a través de bases de dades d'ESTs i comparacions amb el genoma de ratolí. Amb les regions codificants més fiables fem una anàlisi de la proteïna que codifiquen mitjançant bases de dades de proteïnes humanes i de dominis proteics.
Finalment hem trobat interessant fer un estudi sobre el contingut de nucleòtids C+G de la seqüència de 2Mb per tal d'averiguar si existeix o no uniformitat en el contingut d'aquests nucleòtids al llarg de tota la seqüència.Per tal de fer-ho hem desenvolupat un petit programa en Perl que ens ha permès contar la proporció d'aquests nucleòtids en finestres de la seqüència per tal de poder representar després en un gràfic les oscil·lacions del contingut de C+G i extreure'n informació. Per la nostra sorpresa, hem trobat una regió de la seqüència on s'observa una devallada significativa que es correspon a la regió del gen CFTR. La hipòtesi que ens plantegem és la d'un fenomen de transposició, és a dir, la regió que surt de la uniformitat de la seqüència pot haver estat insertada des d'un altre cromosoma a través d'un transposó. No obstant, observant les repeticions emmascarades pel programa RepeatMasker no observem cap increment de les repeticions corresponents a les que acompanyen els transposons, tot i que hem trobat bibliografia que parla d'aquest fenomen en la seqüència que estudiem.
Índex
INTRODUCCIÓ
Les seqüències genòmiques tal i com les obtenim per seqüenciació no ens donen cap informació, cal un estudi esxhaustiu sobre aquestes seqüències per tal de trobar els element funcionals i poder comprendre el funcionament d'aquell genoma. El projecte Encode (ENCyclopedia Of DNA Elements) creat per The National Human Genome Research Institute (NHGRI) pretén identificar tots els elements funcionals en la seqüència del genoma humà. Aquest projecte es troba en la seva fase pilot on es pretén anotar intensament 44 regions escollides a l'atzar o pel seu interès biològic i que corresponent a l'1% del genoma humà. D'aquesta manera es vol testar els diferents mètodes que existeixen per fer una anàlisi rigorosa d'aquestes regions.
En el nostre treball volem buscar i analitzar aquells elements funcionals d'una de les regions de la fase pilot del projecte Encode, en concret, la regió CFTR que es troba al ccromosoma 7 (també anomenada ENm001). Aquesta regió com el seu nom indica comprèn el gen CFTR que codifica per la proteïna reguladora de la conductància transmembrana de la fibrosis quística. Aquest gen quan es troba mutat en certs nucleòtids és el causant de la malaltia coneguda com la fibrosi quística.
Índex
MATERIALS I MÈTODES
- Obtenció de la seqüència
- Emmascarament de la seqüència
- Predicció de gens ab-initio
- Validació de les prediccions:
- Comparació de la seqüència contra la base de dades d'ESTs
- Comparació de la seqüència amb la regió sintènica de ratolí
- Anàlisi de les proteïnes predites
- Anàlisi del contingut de G+C de la seqüència
- Informe realitzat durant l'elaboració del treball
Hem obtingut la seqüència corresponent a la regió ENm001 del projecte ENCODE utilitzant el servidor UCSC Genome Browser, fent cerca en aquesta pàgina amb els passos:
Regions > ENm001 > DNA > Get DNA
Aquesta regió es troba en el cromosoma 7 i té les coordenades següents: 115.404.472 - 117.281.897
La seqüència està en format fasta i la guardem com a sequencia.fa. Per tal de contar el número de nucleòtids i la proporció de C+G de la seqüència cal passar la seqüència en format tabular. Utilitzarem la següent comanda:
awk '{printf $1}' sequencia.fa > sequencia.tbl
Cal posar una tabulació entre nom de la sequencia i la sequencia en el fitxer sequencia.tbl0.
Per tal d'obtenir el número de nucleòtids de la seqüència ens servim de la següent comanda:
awk '{print length($2)}' sequencia.tbl0
Per contar el contingut de C+G a tota la sequencia ho fem amb la comanda:
awk '{print$2}' sequencia1.tbl0 | fold -1 | \
sort | uniq -c | gawk '{print $2, $1/1877426}'
Els programes computacionalment intensius que utilitzarem, com són el Megablast, el RepeatMasker o el TBLASTX de l'SGP2, no permeten seqüències de la llargada de la seqüència que analitzem (1877426 nucleòtids).Així doncs,cal obtenir subseqüències de la seqüència. Per tal de tallar la seqüència en subseqüències solapants ens cal el programa fastachunk. Cada subseqüència serà de 350000 nucleòtids i els fragments solapants seran de 50000. Cal exportar el programa fastachunk. L'exportem amb la comanda:
export PATH=$PATH:/disc8/bin
Per executar el programa fastachunk utilitzem la comanda:
( echo ">subseq1"; fastachunk sequencia.fa 0 350000 | fold -60 ) > subseq1.fa
( echo ">subseq2"; fastachunk sequencia.fa 300000 350000 | fold -60 ) > subseq2.fa
( echo ">subseq3"; fastachunk sequencia.fa 600000 350000 | fold -60 ) > subseq3.fa
( echo ">subseq4"; fastachunk sequencia.fa 900000 350000 | fold -60 ) > subseq4.fa
( echo ">subseq5"; fastachunk sequencia.fa 1200000 350000 | fold -60 ) > subseq5.fa
( echo ">subseq6"; fastachunk sequencia.fa 1500000 377426 | fold -60 ) > subseq6.fa
Taula 1
Emmascarem les subseqüències a través del programa Servidor EMBL del Repeat Masker.
Per a cada bseqüència obtenim els següents 3 fitxers:
Taula 2
Cal treure els residus del format html, ho fem amb la comanda:
egrep -v '^[ \t]*$|^[ \t]*<' subseq1.fa.msk > subseq1fasta.fa.msk
Taula 3
Utilitzem el programa gff2ps per tal de visualitzar el contingut i distribució de les repeticions obtingudes. Cal exportar aquest programa amb la comanda:
export PATH=/disc8/soft/perl/bin/:/disc8/bin/:$PATH
Abans de tot, les subseqüències dels fitxers subseq().repeatmasker.out s'han de passar al format gff amb la següent comanda awk:
grep subseq1 subseq1.repeatmasker.out | \
awk 'BEGIN{ OFS="\t" }
{ print $5, $11, "repeat", $6, $7, ".", ".", "."; }
' > subseq1.repeatmasker.out.gff
Taula 4
Per fer anar el programa gff2ps cal posar la comanda:
gff2ps subseq1.repeatmasker.out.gff > subseq1.repeatmasker.out.ps
Taula 5
Convertim el format ps que acabem d'obtenir a una imatge de bits en format png amb la comanda:
convert -antialias -rotate 90 subseq1.repeatmasker.out.ps subseq1.repeatmasker.out.png
Taula 6
Utilitzem 3 programes de predicció ab-initio per predir els gens al llarg de la seqüència. Utilitzarem els següents programes:
- GENEID:
Utilitzem el Geneid local cridant-lo a la terminal i l'executem. Primer per obtenir un fitxer en format gff i després per obtenir un fitxer out.
Per obtenir el fitxer amb el format gff utilitzem la següent comanda per cridar el programa i transformar el fitxer subseqxfasta .fa a gff:
geneid -G -P /disc8/bin/geneidparams/human3iso.param \
subseq1fasta.fa.msk > subseq1geneid.fa.gff
Taula 7
Per obtenir el fitxer amb el format out utilitzem la següent comanda per cridar el programa i transformar el fitxer subseq()fasta .fa a .out:
geneid -D -P /disc8/bin/geneidparams/human3iso.param \
subseq1fasta.fa.msk > subseq1geneid.fa.out
Taula 8
- GENSCAN:
Utilitzem el genscan local a la terminal.Amb la següent comanda obtenim un fixer out que cal passar a format gff amb una altra comanda.
genscan /disc8/bin/genscanparams/HumanIso.smat \
../../masksubseq1/subseq1fasta.fa.msk -cds \
> subseq1.genscan.out
Taula 9
Per passar-ho a format gff ho fem amb la comanda:
gawk 'BEGIN{OFS="\t"}
$2 ~ /Term|Intr|Init/ {
print "subseq1", "genscan", $2, start=($4<$5 ? $4 : $5),
end=($5<$4 ? $4 : $5), $13, $3,
$7, $1;
}' subseq1.genscan.out | \
sed 's/\.[0-9][0-9]$//' > subseq1.genscan.out.gff
Taula 10
- FGENESH :
El programa FGENESH l'hem utilitzat des d'un servidor web. El fitxer dels resultats obtinguts l'hem obtingut en format text i en format HTML:
Taula 11
Taula 12
Cal obtenir el format gff a través del fitxer de text obtingut amb la comanda:
gawk 'BEGIN{OFS="\t"}
$4 ~ /CDSf|CDSi|CDSl/ {
print "subseq1", "fgenesh", $4, start=($5<$7 ? $5 : $7),
end=($7<$5 ? $5 : $7), $8, $2, $3, $1;
}' subseq1.fgenesh.out | \sed 's/\.[0-9][0-9]$//' >
subseq1.fgenesh.out.gff
Taula 13
A continuació,per tal d'observar els resultats de les prediccions realitzades pels 3 programes generem un gr`fic mitjançant el programa gff2ps. Ho fem a través de la següent comanda:
gff2ps predicciogens/geneid/subseq1geneid.fa.gff\
predicciogens/fgenesh/subseq1.fgenesh.out.gff\
predicciogens/genescan/subseq1.genscan.gff > subseq1.graficpre.ps
Taula 14
Convertim el format ps a una imatge de bits en format png amb la comanda:
convert -antialias -rotate 90 subseq1.graficpre.ps subseq1.graficpre.png
Taula 15
- Comparació de la seqüència contra la base de dades d'ESTs
Per tal de validar la predicció feta pels programes de predicció ab-intio compararem la nostra seqüència contra la base de dades d'ESTs humans utilitzant el programa Mega BLAST , del servidor NCBI BLAST.
En el programa MegaBLAST em deixat per defecte tots els paràmetres excepte:
- Base de dades : est-human
- Deseleccionar: Graphical Overview, Linkout i Sequence Retrieval
- Descriptions: 1000 i Alignments:1000
- Alignment view: pairwise
- Layout: one window
- Formatting options on page with results:at the bottom
Els resultats del MegaBLAST els guardarem en format text i en format HTML:
Taula 16
Taula 17
Per tal de passar el fitxer de text a format gff cal primer de tot eliminar completament les restes de format HTML que hi queden. Ho fem amb la comanda:
perl -ne 's/\>/>/og; # recuperem el caracter ">"
s/\&.+?;//og; # treiem qualsevol altre caracter codificat en HTML
s/\<.+?\>//og; # treiem tots els tags de HTML
s/^-->//o; # eliminem el que queda davant de "*BLAST*"
$. > 4 && print; # i ens salten les 4 primeres linies que tambe son HTML
' subseq1.megablast.txt > subseq1.megablast.out
Taula 18
Ara ja podem passar el resultat a format gff, mitjançant el programa parsemegablast.pl. Primer cal exportar el programa amb la comanda:
exportMEGABLAST2GFF="/home/u21439.est12.alu.upf/novell/home/treball/megablast/parsemegablast.pl"
Executem el programa de la manera següent:
perl $MEGABLAST2GFF -G subseq1.megablast.out > subseq1.megablast.gff
Taula 19
A continuació cal FILTRAR els ESTs per trobar aquells que estiguin suportats per splicing.
Utlilitzarem el programa getsplicedhsp.awk per obtenir els Spliced ESTs.
Executem el programa sobre els nostres fitxers en format gff:
gawk -f getsplicedhsp.awk subseq1.megablast.gff > subseq1.megablast.spliced.gff
Taula 20
Després cal ELIMINAR EL FRAME(sentit) dels ESTs perquè en el gràfic posterior no ens separi els ESTs de diferents sentits.
Utilitzem la següent comanda gawk:
gawk '{$7=".";print $0}' subseq1.megablast.spliced.gff > subseq1.megablast.splicednotframe.gff
Taula 21
GRÀFIC CONJUNT DE LES PREDICCIONS AB-INITIO I RESULTAT DE MEGABLAST
Per tal de fer un gràfic conjunt amb les prediccions ab-initio i els ESTs trobats, filtrats i sense frame del megablast tornem a utilitzar el programa gff2ps.
La comanda que utilitzem per obtenir el gràfic conjunt és:
gff2ps predicciogens/geneid/subseq1geneid.fa.gff \
predicciogens/fgenesh/subseq1.fgenesh.out.gff \
predicciogens/genescan/subseq1.genscan.out.gff \
megablast/subseq1.megablast.splicednotframe.gff > subseq1.validacio.ps
Taula 22
Convertim el format ps a una imatge de bits en format png amb la comanda:
convert -antialias -rotate 90 subseq1.validacio.ps subseq1.validacio.png
Taula 23
- Comparació de la seqüència amb la regió sintènica de ratolí
Per tal d'acabar de validar les nostres prediccions hem utilitzat el programa SGP2, que ens alinia dues seqüències homòlogues, en el nostre cas la d'humà i la de ratolí, i ens prediu segons la conservació dels nucleòtids, les regions que són codificants. Les regions que analitzem amb aquest programa són aquelles regions codificants predides pels 3 programes de predicció de gens i que estan més validades per ESTs.
Una vegada escollides les regions, hem d'obtenir les seqüències de nucleòtids d'aquestes regions que passarem en el programa d'alineament BLAT , per tal d' obtenir després les regions sintèniques en el ratolí.
Les regions que escollim contenen el gen predit i almenys un miler de bases per davant i un miler per enrera per tal de trobar la regió que busquem en el ratolí.
El que cal tenir present també és que aquelles regions que ens superin les 100Kb no les analitzarem ja que el programa SGP2 només accepta regions menors de 100Kb.
Així doncs a través altra vegada del programa fastachunk hem realitzat les següents comandes per a obtenir la regions que corresponen als gens (predit per els programes ab-initio).
Taula 24
No obstant, el programa BLAT no admet més de 25000 nucleòtids, així doncs, cal utilitzar el programa fastachunk per tallar cada regió escollida en fragments de 25000 o menys nucleòtids. Podeu accedir a les comandes realitzades amb el fastachunk a través del link comandes.
Taula 25
BLAT
Una vegada tenim les regions dividides per tal de poder fer l'aliniament amb el BLAT anem al servidor que ens proporciona el programa i canviem els següents paràmetres:
- genome: mouse
- query type: DNA
- sort output: chrom,score
Quan decidim quina regió del genoma del ratolí ens quedem per posteriorment utilitzar el programa SGP2 podem exportar la regió des del mateix servidor web del BLAT. Posem les coordenades de la regió del genoma del ratolí que volem i triem l'opció "Get DNA". Triem la opció emmascarar amb N. Guardem aquesta seqüència sota el nom de:
Taula 26
Programa SGP2
Farem correr el programa SGP2 dues vegades per cada regió per poder obtenir els resultats en dos formats:
Seleccionant
Output format:GFF
obtenim un fitxer en format gff. Els fitxers en format gff es resumeixen a la taula:
Taula 27
Seleccionant
Output format:geneid including CDS sequence
obtenim un fitxers en format .txt resumits en aquesta taula:
Taula 28
El gen 2 de la subseqüència 5 correpon al gen CFTR. Tot i que les seqüències tant de ratolí com d'humà són superiors a 100,000 bases ens interessa poder analitzar amb aquest programa la predicció de regions codificats, així és que haurem de tallar les dues seqüències. Ho farem com sempre utilitzant el programa Perl fastachunk.
Tallem la humana ( 2 fragments de 95,000 amb solapament de 10,000 i l'ultim fragment de 90,000) utilizant les següents comandes i guardem els fragments en els següents fitxers:
Taula 29
Tallem la de ratoli ( 3 fragments solapants, els dos primers de 70,000 i l'ultim de 75,000) amb les següents comandes i guardem els fragments en els següents fitxers:
Taula 30
Passem la primera humana amb la primera de ratoli per el SGP2. EL mateix per el segon i el tercer framgmet.Així obtenim dos fitxers de cada tall fet en la regió del gen2 de la subseqüència 5:
- huma_ratoli.subseq5_gen2aCDs.txt
- huma_ratoli.subseq5_gen2a.gff
- huma_ratoli.subseq5_gen2bCDs.txt
- huma_ratoli.subseq5_gen2b.gff
- huma_ratoli.subseq5_gen2cCDs.txt
- huma_ratoli.subseq5_gen2c.gff
GRÀFIC CONJUNT DE LES PREDICCIONS AB-INITIO, ESTS I REGIONS SINTÈNIQUES DEL RATOLÍ:
Per a fer la gràfica de la predicció, juntament amb els resultats del SGP2, primer cal passar les coordenades de la regió genòmica a les coordenades absolutes de la subseqüència que estem treballant mitjançant les següents comandes gawk:
Taula 31
Ara ja podem crear el gràfic conjunt utilitzant el programa gff2ps. La comanda que fem servir la modifiquem de les utilitzades anteriorment per tal que en aquest gràgic els ESTs es representin en una sola linia al gràfic.
Així obtenim el format ps de cada subseqüència:
Taula 32
Pel cas de la subseqüència 5, que conté un gen predit per SGP2 dividit en tres regions primer de tot utilitzarem la següent comanda per tal d'ajuntar les prediccions de les tres regions del gen 2 i així mostrar-les en una sola linia al gràfic:
cat sintenica/SGP2/huma_ratoli.subseq5_gen2a.absolutes.gff
sintenica/SGP2/huma_ratoli.subseq5_gen2b.absolutes.gff
sintenica/SGP2/huma_ratoli.subseq5_gen2c.absolutes.gff >
sintenica/SGP2/huma_ratoli.merged_sgp.absolutes.gff
La següent comanda és la que usem per produir el fitxer ps.
Ens sembla interessant afegir al gràfic les coordenades reals del gen CFTR. Per tal d'obtenir-les usem la base de dades UCSC Genome Browser on Human May 2004 Assembly i ho guardem en format gff sota el nom: CFTR.gff .
Del fitxer en format gff hem de quedar-nos només amb les coordenades del CDs,això ho fem amb una comanda grep:
grep CDS CFTR.gff >CFTR.grep.gff
Per tal que les coordenades trobades, que son absolutes del cromosoma,
siguin relatives a la nostra subseq5 que es on hem trobat nosaltres el
gen cal fer una comanda gawk. La nostra sequencia de 2000000 de bases
comença pel que fa a coordenades absolutes del cromosoma en 115404472,
i la subseq5 respecte la sequencia que estudiem a 1200000.
Per tant, per dur a coordenades relatives a subseq5 el gen real cal:
115404472+1200000=116604472 i aixo restar-ho a les coordenades
absolutes del gen real:
gawk ' BEGIN{OFS="\t"}{ $4 = $4 - 116604472 ; $5 = $5 - 116604472 ;
print }
' CFTR.grep.gff > CFTR.grep_absolutes.gff
Afegim les coordenades del gen CFTR reals en el gràfic seguint les següent comanda del link
Convertim el format ps de tots els gràfics que hem obtingut en aquest apartat a una imatge de bits en format png amb la comanda:
convert -antialias -rotate 90 validacio+SGP2.subseq2_gen1+2_collapsedESTs.ps\
validacio+SGP2.subseq2_gen1+2_collapsedESTs.png
Taula 33
Utilitzem el programa de l'NCBI BLASTP per comparar la predicció de la proteïna dels gens predits contra una base de dades per tal d'identificar-les. Hem deixat per defecte tots els paràmetres del programa excepte:
Choose database: SWISSPROT
select from: Homo sapiens
Layout: One window
Formatting options on page with results: at the bottom
La pàgina web de resultats l'hem desada amb el nom:
Taula 34
Utilitzem la base de dades InterPro per tal d'obtenir els dominis funcionals que conté la proteïna predita. Ens guardem la pàgina web dels resultats sota el nom de:
Taula 35
Per tal de fer l'estudi sobre el contingut de nucleòtids C+G al llarg de la nostra seqüència hem ideat un petit programa en Perl que l'hem anomenat: contargc.pl.
Aquest programa ens permet obtenir; la proporció de C+G dins finestres lliscants que recorrent la seqüència. La longitud de la finestra és un paràmetre escollit per l'usuari.
En el nostre cas crearem 5 fitxers que contindran el contingut de C+G de finestres de longitud 500, 1000, 2000, 5000 i 10000 nucleòtids cada un respectivament. La comanda que utilitzem és:
perl contargc.pl 500 sequencia.fa > sequencia.gccontent_500.tbl
perl contargc.pl 1000 sequencia.fa > sequencia.gccontent_1000.tbl
perl contargc.pl 2000 sequencia.fa > sequencia.gccontent_2000.tbl
perl contargc.pl 5000 sequencia.fa > sequencia.gccontent_5000.tbl
perl contargc.pl 10000 sequencia.fa > sequencia.gccontent_10000.tbl
A partir del paquet estadístic R obtenim un gràfic que ens mostra de manera visual les oscil.lacions de C+G al llarg de la seqüència que integra els tres fitxers anteriors.
Exportem aquest programa amb la comanda:
export PATH=$PATH:/disc8/soft/R/bin
A través de la següent comanda obtenim el gràfic que integra els fitxers resultat de programa contargc.pl. A més, també afegim al gràfic un indicador de la localització de la subseqüència 5 i ressaltem el gen CFTR amb una caixa de color blau.
D'aquesta manera obtenim el gràfic sequencia.gccontent_combo.png
Per tal de centrar-nos en la subseqüència 5 passem el programa contargc.pl en longitud finestra 500:
perl contargc.pl 500 subseq5.fa > subseq5.gccontent_500.tbl
I ara, farem el gràfic utilitzant R, però de tal manera que ens indiqui el gen CFTR i els seus exons dins el gràfic.
Primer utilitzarem una comanda gawk per seleccionar les coordenades inici i final de cada exò del gen CFTR i el fitxer resultant el passarem per el programa R, juntament amb la subseqüència 5 i les coordenades del gen CFTR. Les comandes que passarem per R estàn indicades en aquest link .El grà resultant l'anomenem sequencia.gccontent_combo_zoom5.png.
Per a més informació podeu consultar a l'informe realitzat durant l'elaboració del treball.
Índex
RESULTATS
- Obtenció de la seqüència
- Emmascarament de la seqüència
- Predicció de gens ab-initio
- Validació de les prediccions:
- Comparació de la seqüència contra la base de dades d'ESTs
- Comparació de la seqüència amb la regió sintènica de ratolí
- Anàlisi de les proteïnes predites
- Anàlisi del contingut de G+C de la seqüència
- Obtenció de la seqüència
A continuació veiem un esquema de la regió cromosòmica de la seqüència que analitzem:

La nostra seqüència té una llargada de 1877426 nucleòtids.
El contingut de cada nucleòtid a la nostra seqüència és:
A: 0.305551
C: 0.1925
G: 0.191955
T: 0.309994
Així és que el contingut en C+G de la seqüència és de: 0.38445.
La seqüència és massa llarga per tal de poder treballar amb els programes que necessitem. Així és que l'hem dividit en 6 subseqüències solapants que podeu veure en la (Taula 1>.
- Emmascarament de la seqüència
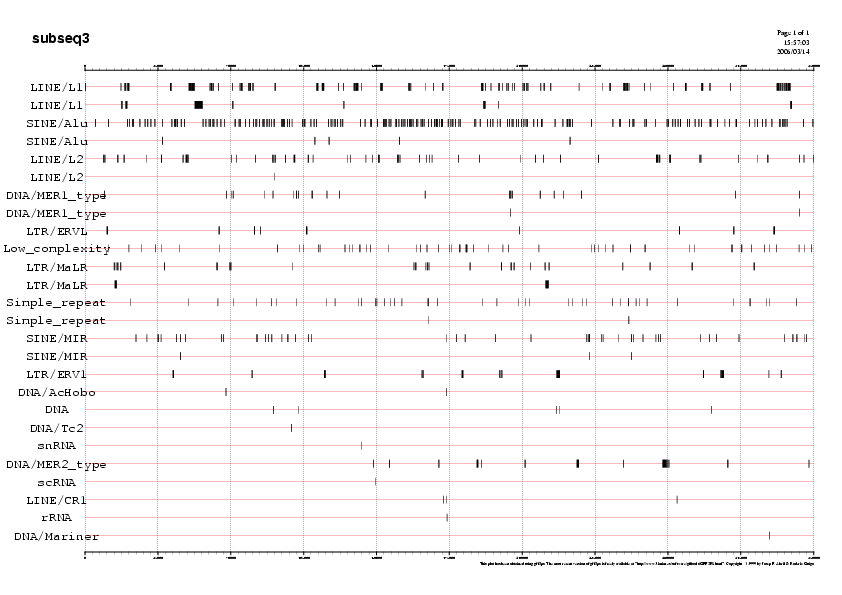
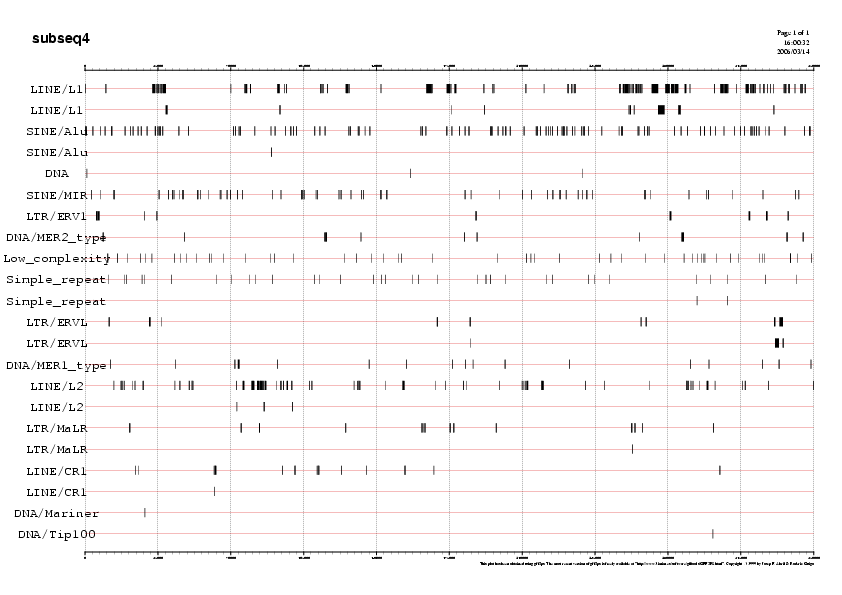
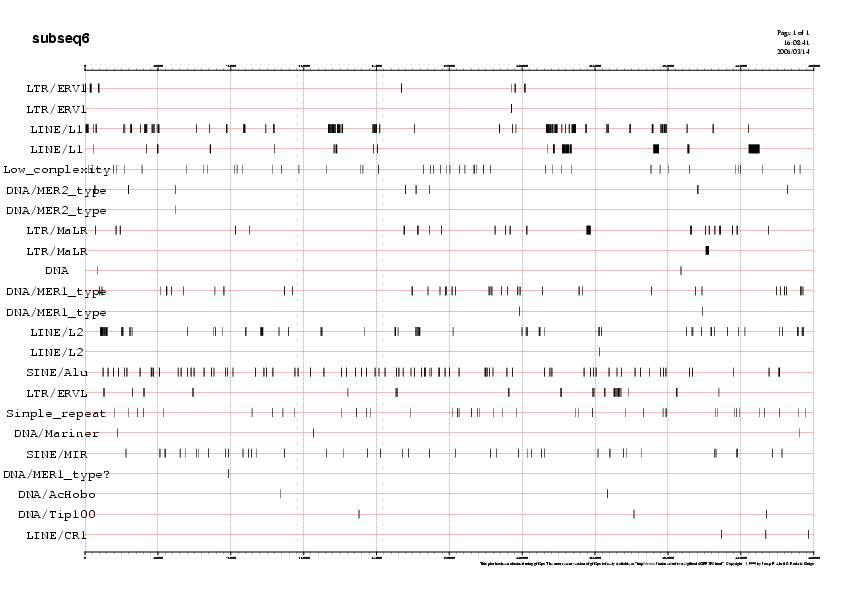
Les taules resum de les repeticions que conté cada subseqüència (Taula 2)ens indiquen uniformitat en la quantitat i tipus de repeticions dins cada subseqüència. Al voltant del 35% de cada subseqüència són elements repetitius i més del 95% d'aquestes respeticions són Interesperced repeats, indicant-nos doncs, que la regió de la seqüència que estudiem no es troba ni en telòmers ni en centròmer.
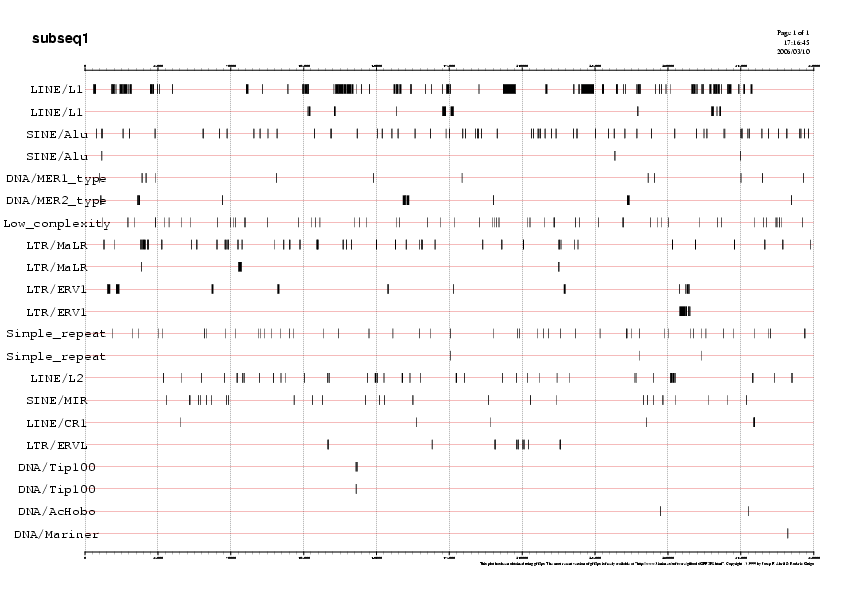
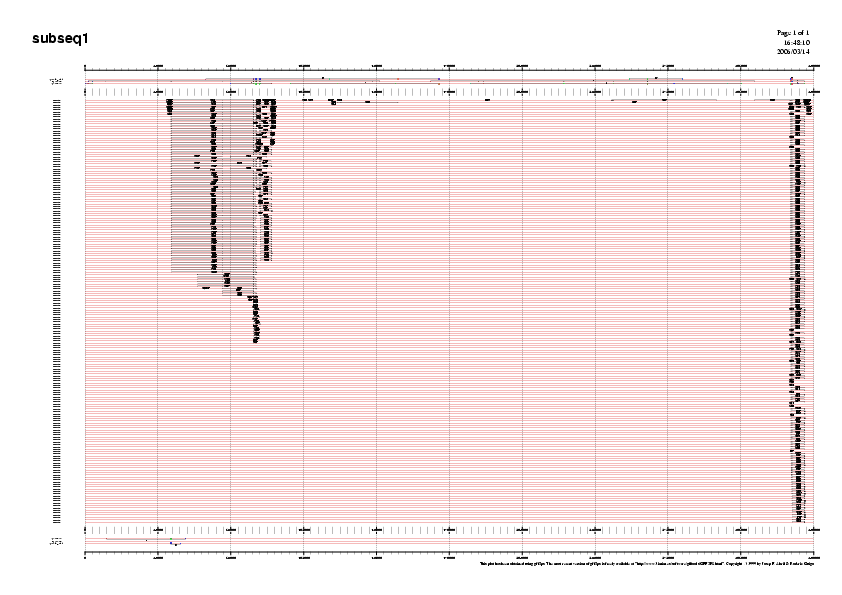
Per tal de visualitzar la distribució de les repeticions al llarg de cada subseqüència podem recòrrer als següents gràfics. Mostrem aquí com a exemple el gràfic de la subseqüència5, ja que és la que analitzarem més exhaustivament:
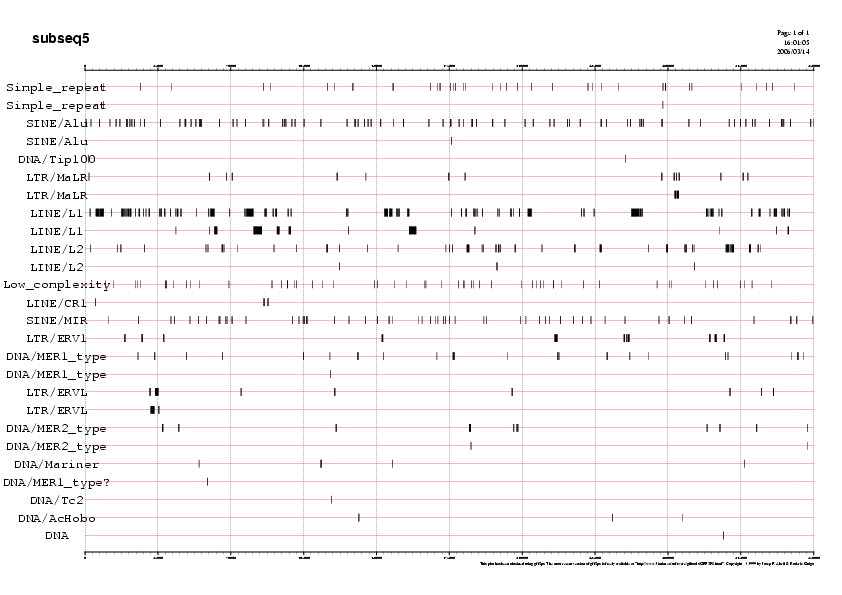
Gràfic 1
Podeu observar la resta de gràfics a la Taula 6.
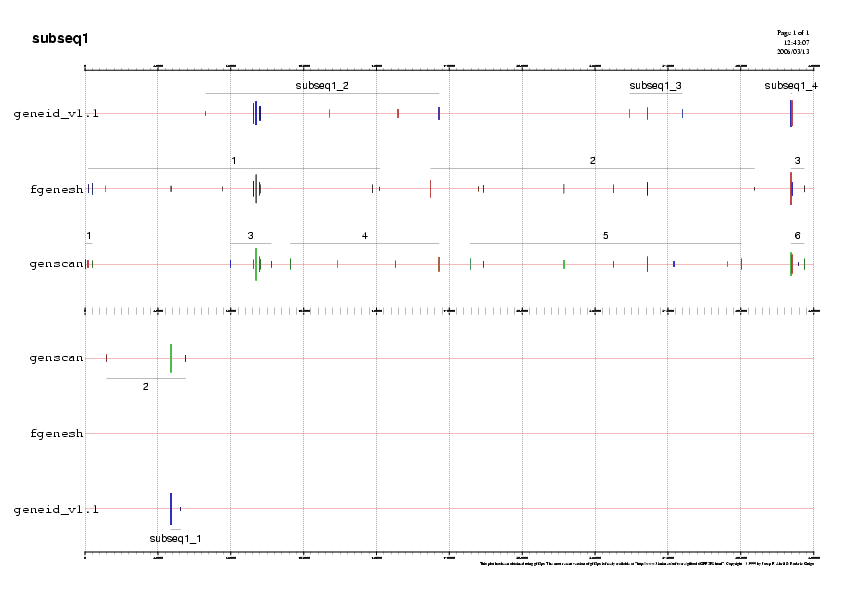
- Predicció de gens ab-initio
Un cop obtingut els resultats dels programes de predicció ab-inito realitzem gràfiques per a poder visualitzar millor les prediccions i les guardem en format png. Podeu observar-les en la Taula 15.
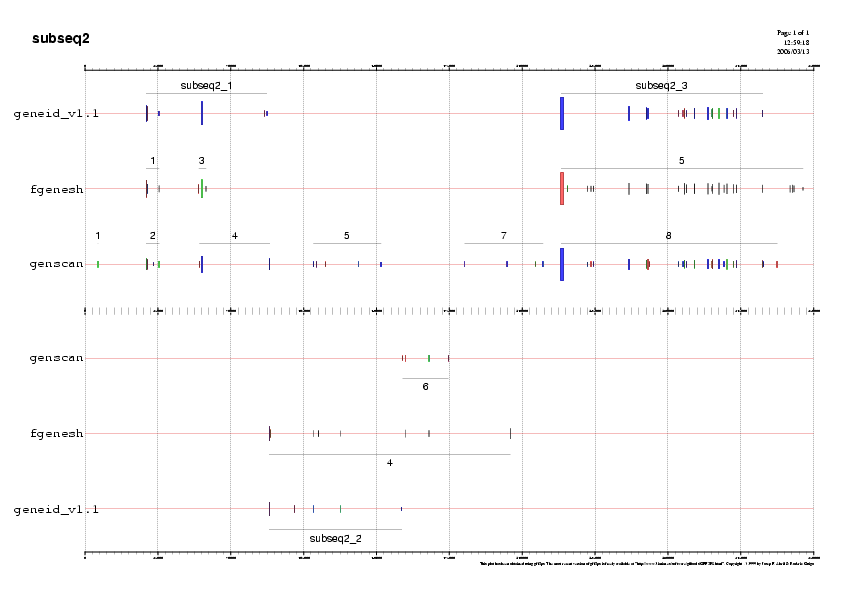
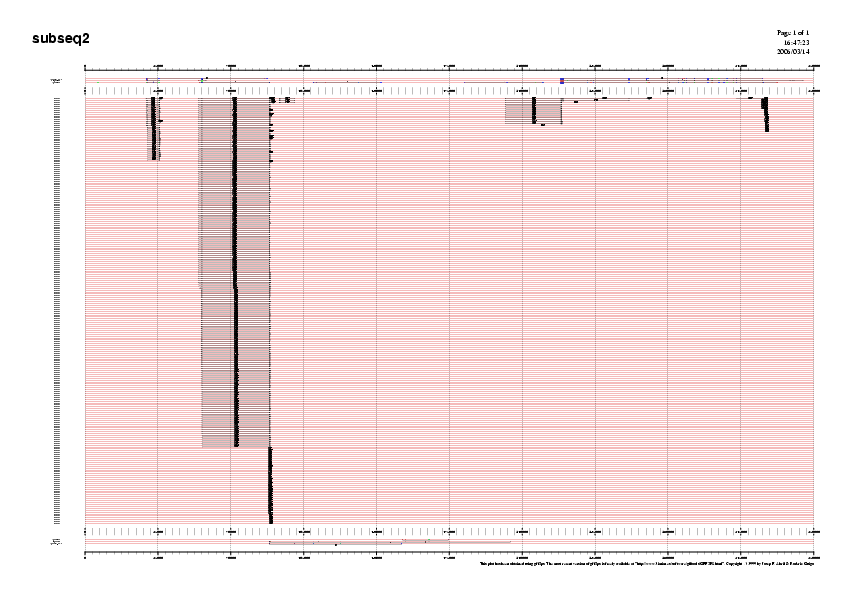
A continuaciò mostrem un exemple d'una gràfica de prediccò, la de la subseqüència 5.
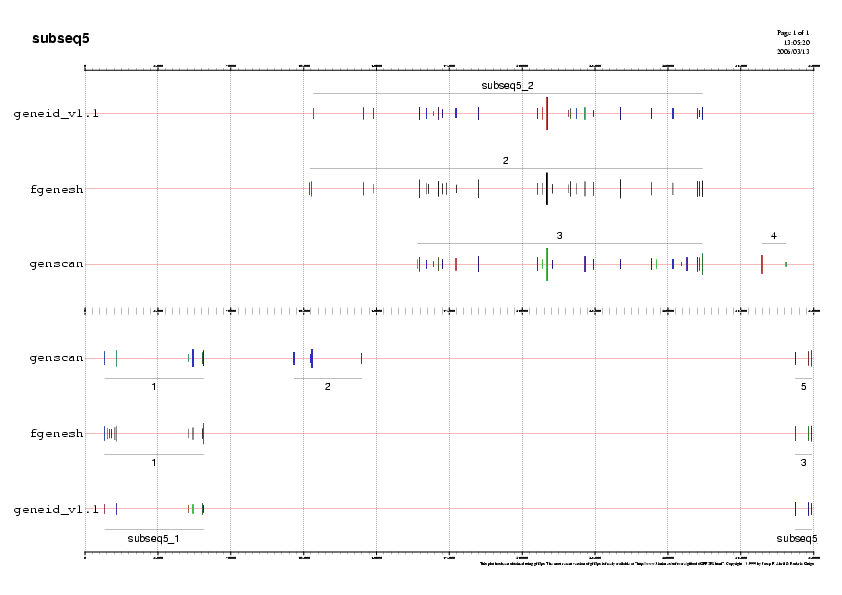
Gràfic 2
- Validació de les prediccions:
- Comparació de la seqüència contra la base de dades d'ESTs
Observant els gràfics dels tres programes de predicció ab-initio juntament amb el ESTs obtinguts del Mega BLAST, podem decidir de cada subseqüència quina de les prediccions de gens pot ser la més encertada.
Degut el gran volum d'ESTs obtinguts ens cal
filtrar-los i organitzar-los per tal de poder extreure'n informació.
- Filtració d'aquells ESTs que estiguin suportats per splicing.
- Eliminació del frame dels ESTs per tal
d'interpretar millor el gràfic.
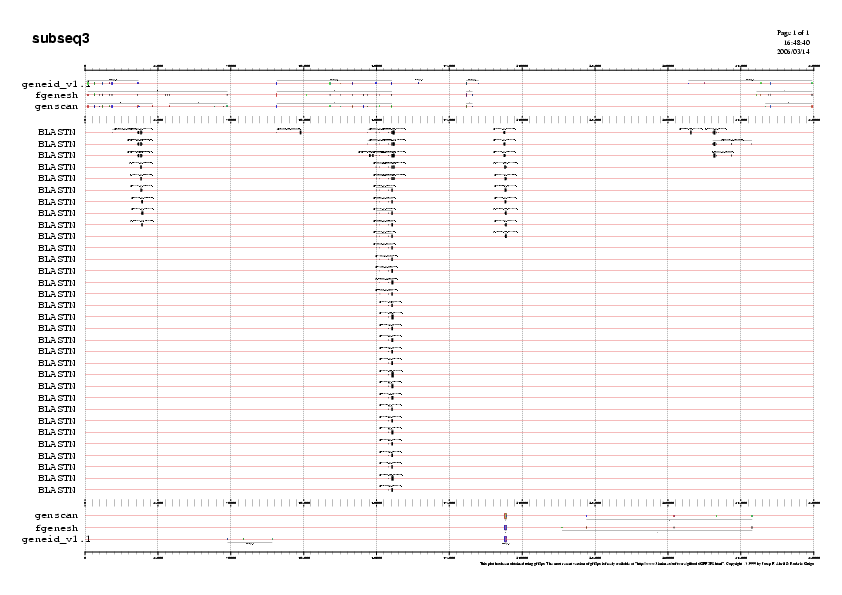
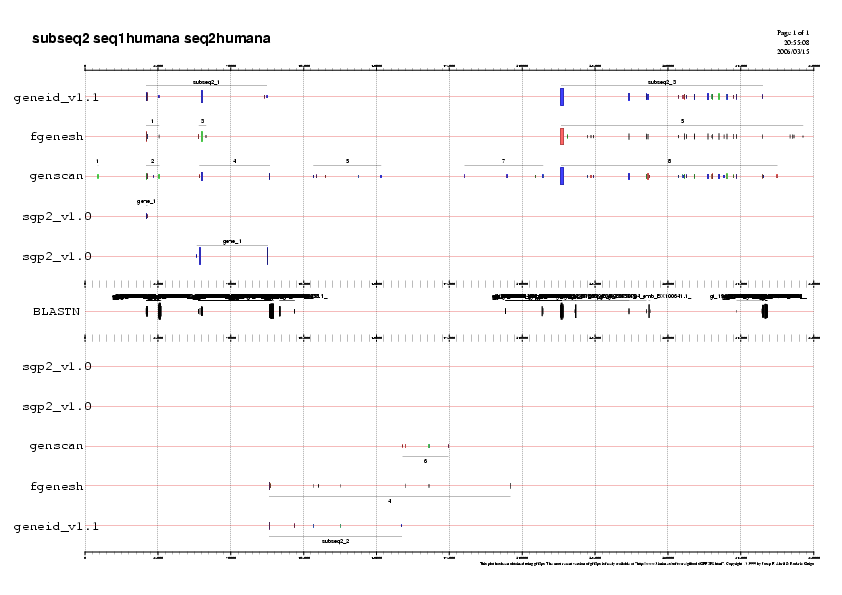
Es pot accedir als gràfics en format png que trobareu a la Taula 23. Aquí mostrem la gràfica de la seqüència5 com a exemple:
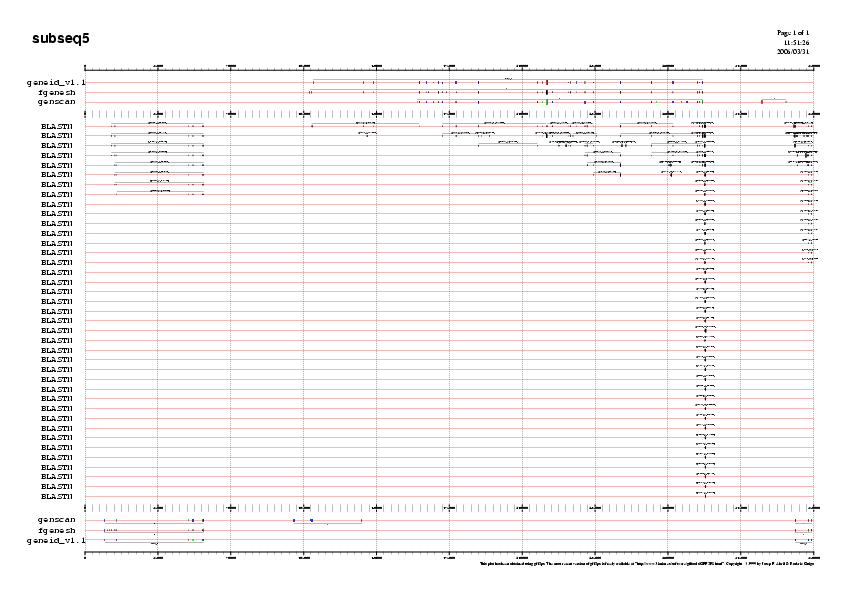
Gràfic 3
A continuació analitzarem subseqüència per subseqüència de 5' a 3'observant els gens predits pels tres programes ab-initio i
seleccionarem aquelles prediccions suportades per ESTs. Aquestes
prediccions les considerarem gens i les etiquetarem amb una lletra.
- Subseqüència 1:
Predicció 1: Els tres programes de predicció ab-initio ens prediuen un gen en forward a la regió 5' de la subseqüència 1.No obstant, les prediccions són molt diferents i per tant, la validació per ESTs ens
pot ajudar a discernir quina de les tres prediccions és millor. Observem
en el gràfic que aquest gen està validat per ESTs
que ens mostren exons que només trobem al gen predit per
Fgenesh. Aquest fet ens fa escollir el gen predit per Fgenesh, serà el
gen A.
Predicció 2: Els tres programes ens prediuen una segona regió
codificant en forward. Però observem que no coincideixen en les
coordenades i no observem cap validació significativa per ESTs.
Predicció 3: Els tres programes ens prediuen una petita regió
codificant a l'extrem 3' de la subseqüència 1 en forward. No obstant,
es pot observar en els fitxers de resultats dels tres programes de
predicció !!!!!!que cap d'aquestes prediccions tenen un exó
terminal. Això ens fa pensar que aquest possible gen ha quedat dividit
entre la subseqüència 1 i la 2. Així doncs, el recuperarem a la subseqüència 2.
- Subseqüència 2:
Predicció 1: Observem que els tres programes de predicció ab-initio
ens prediuen una regió codificant en forward a la regió 5' de la
subseqüència 2. No obstant, les prediccions són molt diferents ja que
per exemple el programa geneid ens prediu un únic gen en aquesta regió
i els altres dos programes, fgenesh i genscan, ens en prediuen dos de
separats. La validació per ESTs ens indica que en aquesta regió hi ha
dos gens separats. Ens quedarem amb la predicció feta per genscan ja
que tant pel segon gen fgenesh no conté tots els exons que ens
indiquen els ESTs. Així dons, d'aquesta regió escollim els dos gens en
fordward que ens prediu genscan. Aquest dos gens els anomenem gen B i
gen C.
Predicció 2: Els tres programes prediuen una regió codificant en
reverse, amb diferents coordenades i que a més no està suportada per
cap EST, així és que la desestimem.
Predicció 3: Genscan prediu un gen en forward que no està validat per
cap EST. També el desestimem.
Predicció 4: Els tres programes prediuen una regió codificant en
forward a l'extrem 3'. No obstant, molts dels exons que ens prediuen
els programes no tenen ESTs que els validin. No ens quedarem amb cap
gen predit pels programes ab-initio en aquesta regió.
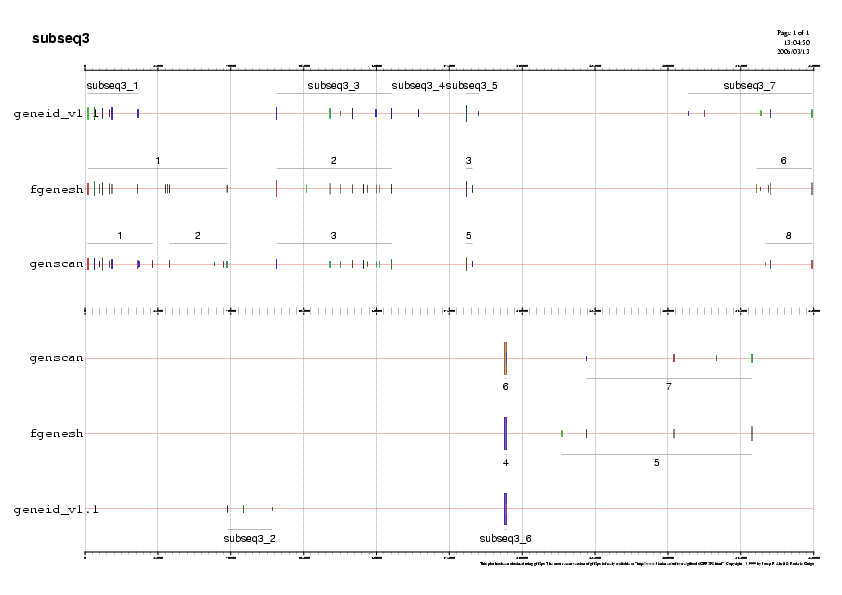
- Subseqüència 3:
Predicció 1: Observem que els tres programes de predicció ab-initio
ens prediuen una regió codificant en forward a la regió 5' de la
subseqüència 3. No obstant, no hi ha consens entre els tres per poder
predir un gen i a més observant els ESTs veiem que només hi ha
cobertura d'aquests per a dos exons de la predicció.Desestimem aquest
gen.
Predicció 2: Geneid és l'únic programa que ens prediu un gen en
reverse i a més no està suportat per cap EST, és a dir que no el
tindrem en compte.
Predicció 3: Observem que els tres programes de predicció ab-initio
ens prediuen un gen en forward i a més trobem consens en les tres
prediccions des del seu inici fins el final. La cobertura d'ESTs és
nombrosa en l'extrem 3' del gen predit. Ens quedem amb la predicció
feta per geneid, ja que aquest programa usa paràmetres específics
d'humans i per tant és més específic.El gen serà el gen D.
Predició 4: Els tres programes prediuen un petit gen en fordward que
no està suportat per cap EST, evidentment el desestimem.
Predicció 5: Els tres programes prediuen un petit gen en reverse que
té dos exons. Observem que aquest gen i els dos exons estan molt
suportats per ESTs. Ens quedem amb la predicció feta per geneid, ja
que aquest programa usa paràmetres específics d'humans i per tant és
més específic. Aquest gen en reverse és el gen E .
Predicció 6: Trobem en la regió 3' de la subseqüència prediccions en forward i en reverse però els pocs ESTs que trobem en aquesta regió prediuen exons que no trobem en els gens predits pels programes ab-initio. No trobem una cooncordança clara per tal d'estimar cap gen.
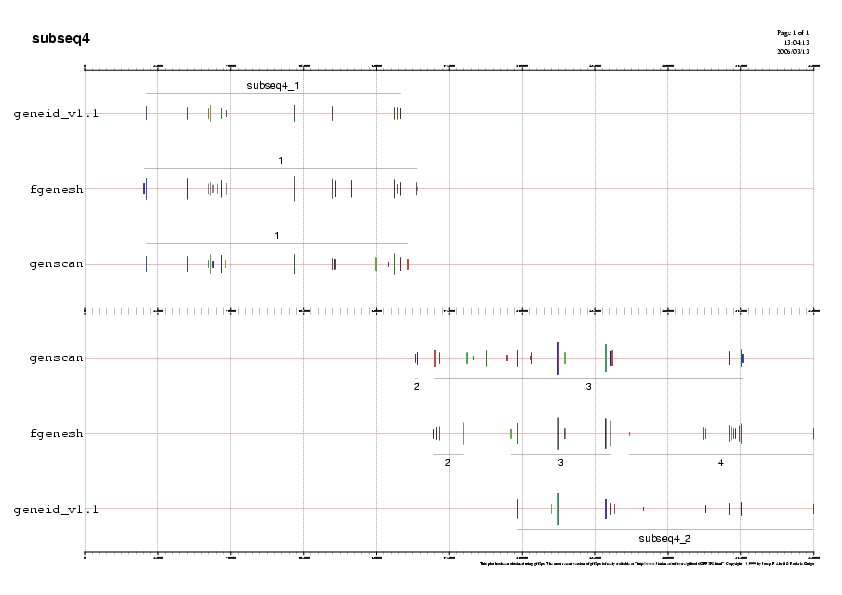
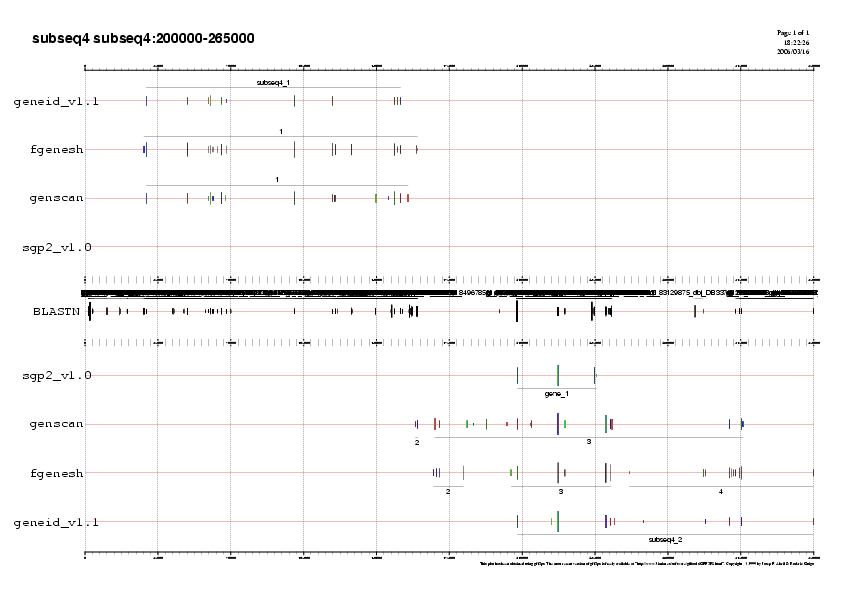
- Subseqüència 4:
Predicció 1: Veiem un gen forward en 5' que està predit per els tres programes ab-initio. Aquestes 3 prediccions estan molt validades per ESTs i per tal de decidir quina prediccióyes ens quedem,analitzem si el exons predits per cada un dels programes està validat per ESTs.
Observem que la predicció del Fgenesh prediu un exóyess en l'extrem 5' i un altre enl'extrem 3',ambdó validats per molts ESTs i no predits per els altres dos programes.
No obstant, si analitzem el fitxer subseq4.fgensh.out, on estan anotats els exons dels gens predits, veiem que no està predit el exó inicial.I per això mateix, la proteïna predita no té una metionina com a primer aminoàcid.
Per tal de decidir si ens quedem amb la predicció de Genscan o Geneid, ens atenem al fet que el programa Geneid segueix uns paràmetres específics humans i per tant serà molt més específic. El gen predit per Geneid li assignem el nom gen F.
Predicció 2: En la següent predicció de la subseqüència, el programa Genscan prediu un gen molt llarg en reverse ( 167500 a 315000). Aquesta predicció no té ESTs en l'extrem 5' ni 3' però si ESTs que validen els exons intermitjos. Com que no hi ha ESTs que cobreixin tota la llargada del gen no acceptem com a valida aquesta predicció.Geneid prediu un gen que està suportat per ESTs, però aquest gen queda partit amb la subseqüècia 5 i no el podem analitzar encara.
Fgenesh prediu 3 gens en aquesta regió. La segon predicció d'aquesta regió, té una cobertura total per ESTs i per tant assignarem a aquesta predicció el nom de gen G.
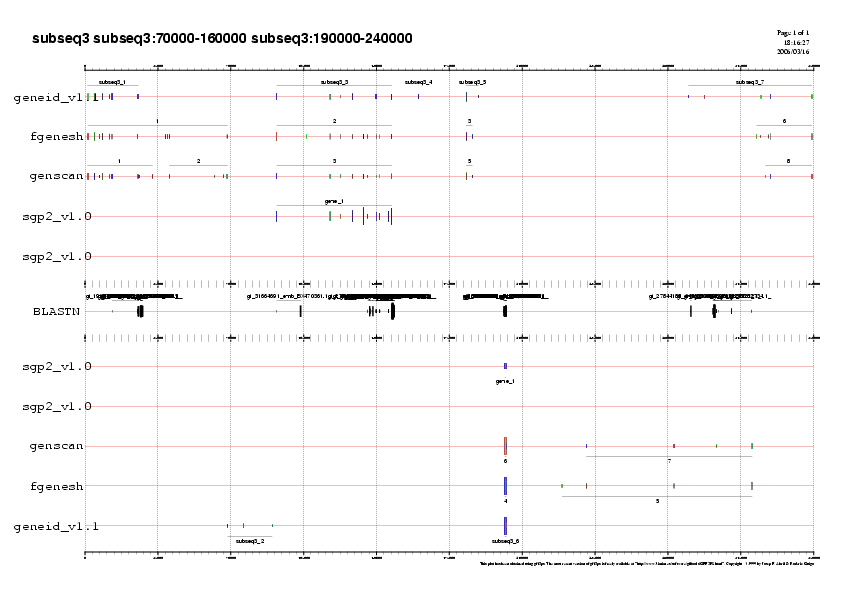
- Subseqüència 5:
Predicció 1: Si observem la primera predicció, veiem que els tres programes prediuen un gen en reverse practicament idèntics en llargada . Aquestes prediccions però presenten diferències en el nombre d'exons predits. La predicció de Geneid no es valida ja que no es prediu l'exo terminal, tal com es pot observar en el fitxer subseq5.geneid.out. Per a decidir amb quina de les dues prediccions restants ens quedem, observem la validació dels exons amb el ESTs. Veiem que els exons predits en 5' són identics en les prediccions de Fgenesh i Genscan i a més estan validats per ESTs. En canvi, els exons 6,7 de la predicció de Fgenesh , no predits per Genscan estan validats per ESTs. Per tan ens quedem la predicció de Fgenesh i li assignem el nom de gen H.
Predicció 2: Les prediccions dels tres programes són molt cooncordants. Els tres programes prediuen un gen amb forward.FGenesh i Geneid prediuen un gen més llarg que Genscan. Com que els exons que prediuen de més Geneid i Fgenesh estan suportats per ESTs,descartem la prediccio de Genscan.
Davant la gran similitud entre la predicció de Geneid i Fgenesh, ens quedem amb la predicció de Geneid, ja que aquest programa ajusta els seus paràmetres a humà i per tant és molt més específic.Anomenarem a aquesta predicció gen I.
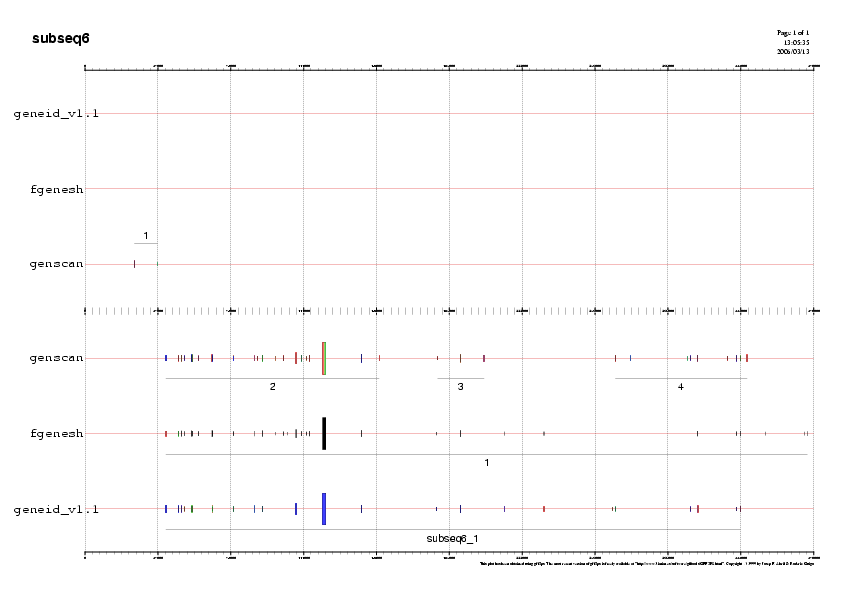
- Subseqüència 6:
Predicció 1: En aquesta subseqüència veiem que els programes geneid i fgenesh prediuen un gen en reverse. Els exons d'aquests gen estan molt suportats per ESTs en l'extrem 3' però no hi ha cap ESTs que validin la regió 5' (aquest biaix és caracterítstic del ESTs).Ens quedem amb la predicció del Fgenesh perquè en el cas del programa Geneid la predicció no té el exó inicial. Anomenarem a a la predicció gen J.
La presència dels ESTs en la regió 5' ens suporta la presència d'un gen sencer enlloc dels 3 gens independents predits per Genscan.
A continuació mostrem una taula resum de les prediccions de tots els gens:
| Subseqüència |
Programa |
Gen |
Exons |
Sentit |
Coordenada inici |
Coordenada final |
| 1 |
Fgenesh |
A |
11 |
+ |
1384 |
137579 |
| 2 |
Genscan |
B |
4 |
+ |
329316 |
335693 |
| Genscan |
C |
3 |
+ |
354935 |
388821 |
| 3 |
Geneid |
D |
6 |
+ |
692146 |
747401 |
| Geneid |
E |
2 |
- |
802445 |
801699 |
| 3-4 |
Geneid |
F |
14 |
+ |
889781 |
1051665 |
| 4 |
Fgenesh |
G |
6 |
- |
1152523 |
1104604 |
| 5 |
Fgenesh |
H |
11 |
- |
1256994 |
1209254 |
| Geneid |
I |
24 |
+ |
1309629 |
1496642 |
| 6 |
Fgenesh |
J |
32 |
- |
1866839 |
1541071 |
Taula 36
Comparació de la seqüència amb la regió sintènica de ratolí
En aquest apartat només obtenim resultats per aquells gens predits que no superin les 100.000 bases, ja que el programa SGP2 només accepta seqüències de menys de 100000 bases. No obstant, el gen I que té una allargada major de 100000 bases el passem pel programa SGP2 en 11 subseqüències solapant. Aquest gen correspon al gen CFTR i ens és interessant poder seguir analitzant-lo.
Així doncs ens quedem amb les següents regions que contenen els gens predits,i que passarem per el BLAT:
| Seqüències que analitzem al BLAT |
| Gen | Coordenades relatives | Coordenades absolutes |
| Inici-Final | Inici-Final |
| B | 5000-40000 | 305000-340000 |
| C | 42000-105000 | 342000-405000 |
| D | 70000-160000 | 670000-760000 |
| E | 190000-240000 | 790000-840000 |
| G | 200000-265000 | 110000 -1165000 |
| H | 5000-70000 | 1205000-1270000 |
| I | 70000-330000 | 1270000-1530000 |
Taula 37
Com es pot observar els gens A, F i J estan en regions que superen les 100000 bases, i per tant no podem passar les seqüències pel BLAT.
BLAT:
El programa Blat de UCSC Genome Bioinformatics Site ens dona la regió sintènica de ratolí de la seqüència que li passem.
Per a cada regió hem hagut d'escollir el hit amb un score i identitat més alts. El que hem pogut observar és que totes les regions sintèniques al ratolí de la nostra seqüència les trobem al cromosoma 6.
En la següent taula presentem els fitxers on es poden observar els regions escollides per cada coordenads de les regions de ratolí que donaven un score més alt al passar les seqüències dels gens per el BLAT. Guardem la regió de ratolí en format fasta.
| Gen | Regió gen humà | Coordenades inici-final | Regió sintènica ratolí |
| B | BLATsubseq2gen1.txt | 17570000-17600000 | mouse_syntenic_ch6.hsap_subseq2_1.fa |
| C | BLATsubseq2gen2.txt | 17240000-17283000 | mouse_syntenic_ch6.hsap_subseq2_2.fa |
| D | BLATsubseq3gen1.txt | 17565000-17610000 | mouse_syntenic_ch6.hsap_subseq3_1.fa |
| E | BLATsubseq3gen2.txt | 17650000-17685000 | mouse_syntenic_ch6.hsap_subseq3_2.fa |
| G | BLATsubseq4gen2.txt | 17925000-17975000 | mouse_syntenic_ch6.hsap_subse4_2.fa |
| H | BLATsubseq5gen1.txt | 18010000-18060000 | mouse_syntenic_ch6.hsap_subseq5_1.fa |
| I | BLATsubseq5gen2.txt | 18090000-18265000 | mouse_syntenic_ch6.hsap_subseq5_2.fa |
Podeu accedir a les regions sintèniques de ratolí amb els linkds de la Taula 26.
Taula 38
- SGP2:
El programa SGP2 ens ha buscat regions codificants a la seqüència aliniant la regió sintènica de ratolí i humà. Els resultats obtinguts queden representats als gràfics següents:
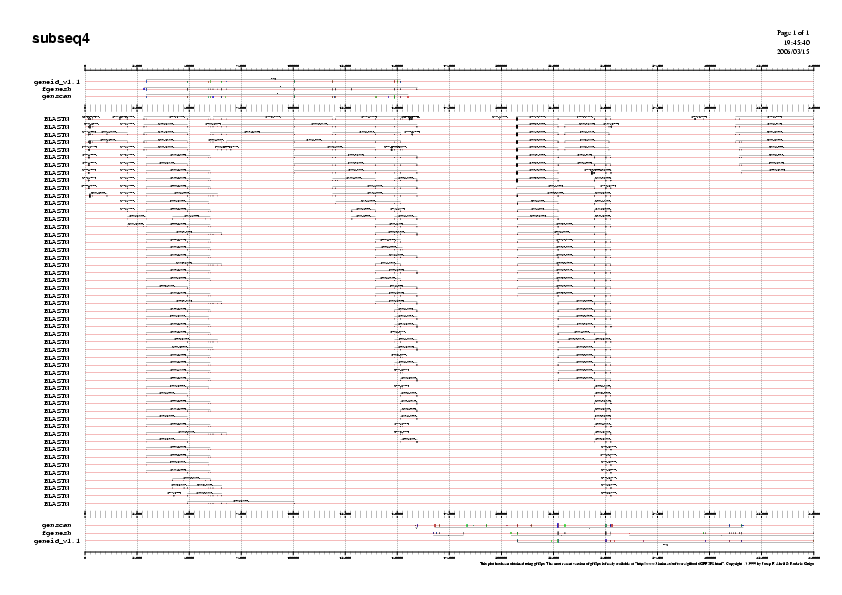
Com a exemple us mostrem el gràic de la subseqüència 5:
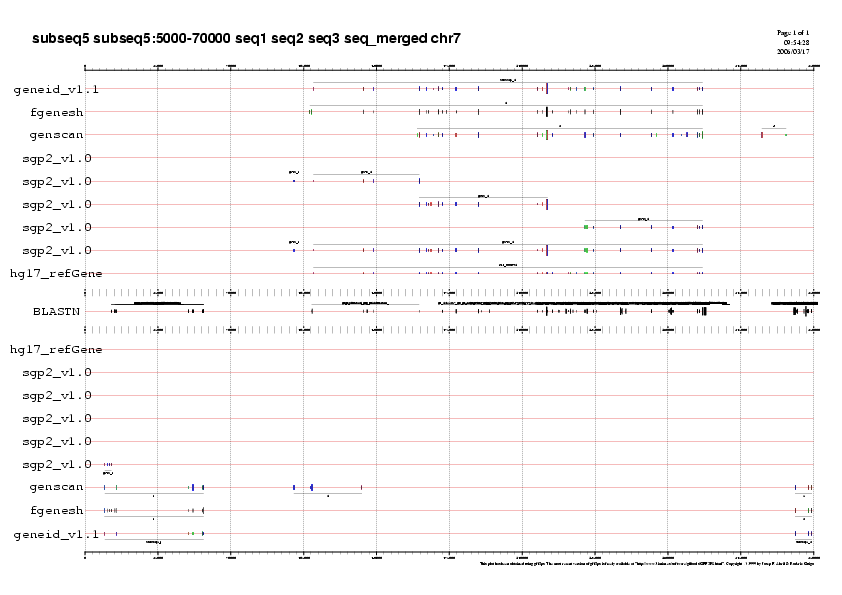
Gràfic 4
Analitzant els gràfics veiem la predicció feta per SGP2 i la podem comparar amb la predicció d'aquells 10 gens que ens hem quedat:
- Gen B i C: Observem que Genscan prediu 4 exons pel gen B, en canvi SGP2 només en prediu 2. Pel que fa al gen C tant Genscan com SGP2 ens prediuen un gen amb els mateixos 3 exons.
- Gen D i E: Veiem quen en el cas del gen D Geneid ens predia només 6 exons, en canvi SGP2 ens en prediu 10. En aquest cas observem que el que més s'acosta a la predicció de SGP2 és Genscan que prediu 9 exons.Pel gen E que es troba en reverse Geneid ens prediu dos exons i SGP2 només un.
- Gen G: Fgenesh ens prediu 6 exons per aquest gen en reverse, SGP2 només en prediu 4, dos d'ells no coincideixen amb cap dels 6 predits per Fgenesh.
- Gen H i I: Fgenesh prediu el gen H amb 11 exons, el programa SGP2 només en prediu els 4 primers. No obstant observem que tres dels quatre exons que prediu SGP2 només es troben predits per Fgenesh i no per cap altre dels dos programes.Pel que fa al gen I corresponen al gen CFTR Geneid ens prediu 24 exons idèntics als que ens prediu SGP2.
Anàlisi de les proteïnes predites
- BLASTP:
A través del programa BlastP per cada una de les proteïnes dels gens predits hem obtingut una sèrie d'aliniaments a la proteïna. Amb aquest programa hem intentat identificar totes les proteïnes.
proteinaA.html
proteinaB.html
proteinaC.html
proteinaD.html
proteinaE.html
proteinaF.html
proteinaG.html
proteinaH.html
proteinaI.html
proteinaJ.html
Taula resum dels resultats:
| Proteïna | aa proteïna | aa proteïna aliniada | aa aliniats | %identitat | Nom de proteïna |
| Proteïna A | 555 | 421 | 358 | 100 | Testin |
| Proteïna B | 219 | 162 | 112>100 | Caveolin-2 |
| Proteïna C | 221 | 178 | 168 | 100 | Caveolin-1 |
| Proteïna D | 152 | 286 | 117 | 61 | F-actin capping protein alpha-2 subunit |
| Proteïna E | 225 | 248 | 246 | 59 | Tropomyosin alpha-4 chain |
| Proteïna F | 463 | * | | | |
| Proteïna G | 378 | 360 | 340 | 69 | Wnt-2 protein precursor |
| Proteïna H | 386 | 475 | 368 | 96 | Ankyrin repeat, SAM and basic leucine zipper domain-containing protein 1 |
| Proteïna I | 1295 | 1480 | 760 | 93 | Cystic fibrosis transmembrane conductance regulator |
| Proteïna J | 1991 | 1773 | 1663 | 79 | Cortactin-binding protein 2 |
Taula 39
*La proteïna F només té un hit, i molt dolent, recordem que aquesta proteïna ve d'un gen que quedava partit entre dues subseqüències. Podria ser que degut a aquest fet el gen estigué mal predit i per tant també la proteïna.
- INTERPRO:
A través del programa Interpro trobem els motius funcionals de les proteïnes predites. Aquest resultat és comparat amb el del BlastP per tal d'observar si hi ha cooncordança entre la proteïna identificada i els motius que s'hi troben.
Els resultats els trobem a:
- Proteina A: Observem que conté dos dominis TESTIN, amb una E-value significativa de 7.5e-225.
- Proteïna B: Observem que conté el domini CAVEOLIN, amb una E-value significativa de 3.7e-81.
- Proteïna C: Observem que també conté el domini CAVEOLIN, amb una E-value també significativa de 1.9e-148.
- Proteïna D: Observem que conté un domini de la subunitat alfa de les proteïnes de la familia F-ACTIN CAPPING. L'E-value del hit és significatiu, 5.1e-29.
- Proteïna E: Observem que el domini que té l'E-value siginificatiu (2.2e-76) és aquell que ens recobreix tota la proteïna, ja que la reconeix com a proteïna de la familia de la tropomiosina.
- Proteïna F: En aquest cas Interpro ens indica la possible identitat de la proteïna F no trobada amb BLASTP. Observem que reconeix en aquesta proteïna la seqüència d'aminoàcids corresponen a les proteïnes de gens de supressió de tumor 7. Té un E-value significatiu de 1.7e-269.
- Proteïna G: El domini que trobem amb un E-value més significatiu (2.6e-170) és WNT RELATED.
- Proteïna H: És una proteïna que conté repeticions Ankyrin, dominis SAM basic leucine zipper.
- Proteïna I: Aquesta proteïna és la del gen CFTR. Observem que conté dos dominis transmembrana tipus ABC amb un E-value significatiu de ABC_tran 3.3e-54 i 8.3e-60.
- Proteïna J: Amb un E-value de 5.4e-120 trobem el domini CORTACTIN-BINDING PROTEIN 2.
Anàlisi del contingut de G+C de la seqüència
El contingut de cada nucleòtid al llarg d'una seqüència que pertany a la mateixa regió genòmica o a un mateix cromosoma sol ser uniforme. La representació gràfica d'acontinuació ens mostra les oscil.lacions del contingut de C+G al llarg de la nostra seqüència. Volem observar si existeix alguna regió on es trenqui aquesta uniformitat. Segons el càlcul realitzat al primer apartat, obtenció de la seqüència, tenim una proporció de nucleòtids C+G de 0,38.
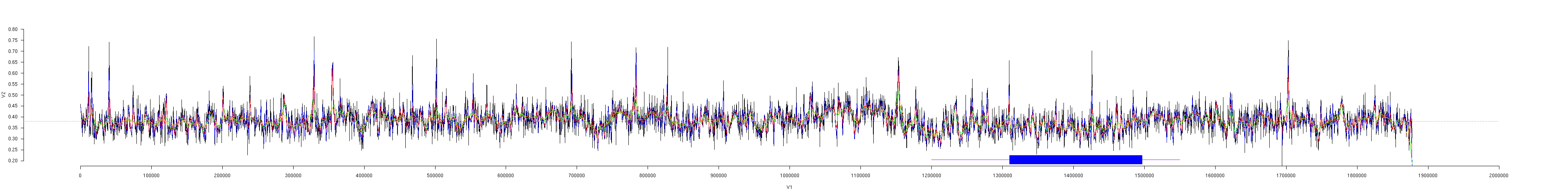
Observem que hi ha una regió de la seqüència on hi ha una baixada pronunciada i sostinguda del contingut de C+G, fins a una proporció de 0,3. Les coordenades d'aquest fenomen coincideixen en les coordenades de la subseqüència 5 i més concretament la zona on trobem el gen CFTR.
Així doncs per tal de visualitzar-ho detalladament podem observar el següent gràfic, on només representem la subseqüència 5:
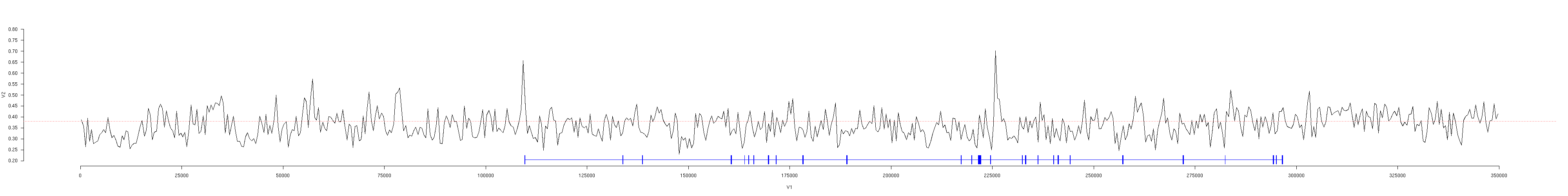 Gràfic 5
Índex
Gràfic 5
Índex
Gràfic 6
DISCUSSIÓ
Tal com hem dit al començar el treball, les seqüències genòmiques tal i com les obtenim per seqüenciació no ens donen cap informació, cal un estudi esxhaustiu sobre aquestes seqüències per tal de trobar els element funcionals i poder comprendre el funcionament d'aquell genoma. A partir de les eines informàtiques que tenim al nostre abast hem intentat complir aquest objectiu.
Els programes de predicció de gens ab-initio ens han fet un primer esbòs de les possibles regions codificants. L'aliniament amb ESTs ens han permès fer una tria d'aquelles prediccions més fiables, en total 10 gens. D'aquí també hem pogut observar quins dels tres programes de predicció ab-initio utilitzats són els més encertats en predir gens en la nostra seqüència. Genscan ens ha predit més gens dels reals, en canvi geneid i fegenesh ens han sigut útils per predir la majoria dels gens suportats per ESTs. Per tal de quedar-nos amb les prediccions més fiables podríem haver utilitzat el programa Exonerate.
El programa SGP2 no ens ha sabut trobar exactament els gens de les regions que li passàvem. Pel que hem observat és un programa que només és útil per validar altres programes de predicció més eficaços que es basen en altres critèris per trobar gens. Després de veure els resultats veiem que hagués sigut interessant utilitzar aquest programa en l'etapa de validació de les prediccions ab-initio, quan decidíem de quin programa ens quedàvem la predicció.
Pel que fa a l'anàlisi del contingut de nucleòtids C+G de la nostra seqüència hem pensat en diferents hipòtesis per tal d'explicar la devallada observada en la regió de la subseqüència 5. Una d'elles és una possible reorganització cromosòmica que afectés a la uniformitat del contingut en C+G, o bé un fenomen de tranposició degut a transposons, fent així, que un tros de seqüència d'un cromosoma saltés a un altre on el contingut de C+G global fos lleugerament superior.
Per tal de veure si la causa és un fenomen de transposició vam tornar a mirar els tipus de repeticions que es trobaven a la regió anterior i posterir de la seqüència amb un contingut de C+G inferior a la mitja. No obstant, no vam poder observar cap increment significatiu de repeticions característiques d'elements transposables com ara LTRs.
Al no poder comprovar les nostres hipòtesis vam buscar referències bibliogràfiques sobre el fenomen de transposició a la regió del gen CFTR. Vam poder correborar que efectivament hi havia grups d'investigació estudiant aquest tema en concret. Un primer article explica que han trobat evidències de transposició en aquesta zona a través de la construcció a partir d'eines informàtiques de la seqüència genòmica ancestral. Un segon article troba aquestes regions a través d'anàlisis comparatius de seqüències multi-espècies de regions genòmiques diana.
- Blanchette M, Green ED, Miller W, Haussler D. Reconstructing large regions of an ancestral mammalian genome in silico.,Genome Res. 2004 Dec;14(12):2412-23. Erratum in: Genome Res. 2005 Mar;15(3):451.[Abstract]
- Thomas JW, Touchman JW, Blakesley, et all.Comparative analyses of multi-species sequences from targeted genomic regions.,Nature. 2003 Aug 14;424(6950):788-93.[Abstract]
Índex
CONCLUSIONS
La finalització d'aquest treball ha permès la identificació de 10 gens i de les seves corresponents proteïnes dins de la seqüència anònima ENm001 de 1.9 Mb, proporcionada per ENCODE. Aques fet ens indica que el protocol estàndard d'anàlisi de seqüències utilitzat és prou adequat tenint en compte la limitació de les eines informàtiques disponibles.
Pensem que seria interessant aprofundir en l'anàlisi de les proteïnes i els seus dominis funcionals trobades per BlastP i Interpro. També seria apropiat i de gran utilitat fer un estudi dels promotors dels gens predits a través del programa Transfac i Promo. No obstant, són estudis que demanen més temps i ens hem vist limitades en aquest aspecte.
Un tema que queda obert i seria interessant i ens agradaria investigar és el fenomen de la baixada pronunciada del contingut de C+G en la regió del gen CFTR.
Índex
REFERÈNCIES
- UCSC Genome Browser:
Kent, W.J., Sugnet, C. W., Furey, T. S., Roskin, K.M., Pringle, T. H., Zahler, A. M., and Haussler, D. The Human Genome Browser at UCSC. Genome Res. 12(6), 996-1006 (2002) [Abstract]
- Fastachunk:
Unpublished program can be provided by Robert Castelo
- Parsemegablast :
Program cedit per cortesia de Josep F.Abril
- Repeatmasker:
A.Smith, unpublished www.genome.washington.edu/uwgc/analysistools/repeatmask.htm ,el qual busca a través d'una acurada base de dades de repeticions utilitzant el programa d'alineament CrossMatch(P.Green, unpublished; http://genome.washington.edu/uwgc/analysistools/swat.htm
- Genscan (local):
Burge, C. and Karlin,S. Prediction of complete gene structures in human genomic DNA J.Mol.Biol.268(1),78-94. (1997) [Abstract]
- Geneid:
R. Guigó, S. Knudsen, N. Drake, and T. F. Smith,
Prediction of gene structure ,
Journal of Molecular Biology, 226:141-157 (1992) [Abstract]
- Fgenesh:
Solovyev, V.V., Salamov, A.A., and Lawrence, C.B. 1995. Identification of human gene structure using linear discriminant functions and dynamic programming. In Proceedings of the Third International Conference on Intelligent Systems for Molecular Biology (ed. C. Rawling et al.), pp. 367-375. AAAI Press, Menlo Park, CA[no electronic reference]
- Mega BLAST:
Zheng Zhang, Scott Schwartz, Lukas Wagner, and Webb Miller. A greedy algorithm for aligning DNA sequences ,J Comput Biol 7(1-2):203-14. (2000) [Abstract]
- BLAT:
W. James Kent. BLAT - The BLAST-Like Alignment Tool ,Genome Res 12:4 656-664 (2002) [Abstract]
- Sgp2:
G. Parra, P. Agarwal, J.F. Abril, T. Wiehe, J.W. Fickett and R. Guigó. Comparative gene prediction in human and mouse. Genome Research 13(1):108-117 (2003) [Abstract]
- BlastP:
Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schäffer,
Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman
,Gapped BLAST and PSI-BLAST: a new generation of
protein database search programs , Nucleic Acids Res. 25:3389-3402.(1997)[Abstract]
- Interpro:
Quevillon E., Silventoinen V., Pillai S., Harte N., Mulder N., Apweiler R., Lopez R
InterProScan: protein domains identifier. Nucleic Acids Research 33: W116-W120 (2005)[Abstract]
- Statistical Data Analysis R
written by Robert Gentleman and Ross Ihaka of the Statistics Department of the University of Auckland. Podeu accedir al programa localment.
Índex
AGRAÏMENTS
Agraïm als professors de l'assignatura l'ajuda i la orientació que ens han permès realitzar aquest treball.
Agraï també molt especialment a les companyes d'anàlisi de seqüència pel suport i les ajudes mútues.
Índex
Si teniu qualsevol dubte podeu contactar amb nosaltres a travès d'e-mail:
Laia Morató i Clara Panosa
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}