Computational Gene Prediction
Practical exercise
|
Abstract: In this exercise, a previously annotated human (HS307871) will be used to
assess the accuracy of different gene finding approaches will be used to annotate
the sequence: ab-initio (GRAIL,
GENSCAN, geneid, FGENESH) and homology-based (GenomeScan, GrailEXP and GENEWISE)
Weak conservation of Start codons will lead
to wrong prediction of initial exons in most cases.
|
There are two main methods for automatic gene prediction: Ab-initio methods and comparative methods:
- Ab-initio methods: they use the DNA sequence as only input. As they use no other information they are usually called
"intrinsic". There are several elements in a gene that can be localized in the genomic sequence,
which are used to identify genes computationally. These elements are related with
the signals that regulate the biological mechanisms of gene expression, and with thesequence bias due to the encoding of a DNA that can be translated into protein. They are called signal sensors and content sensors, respectively.
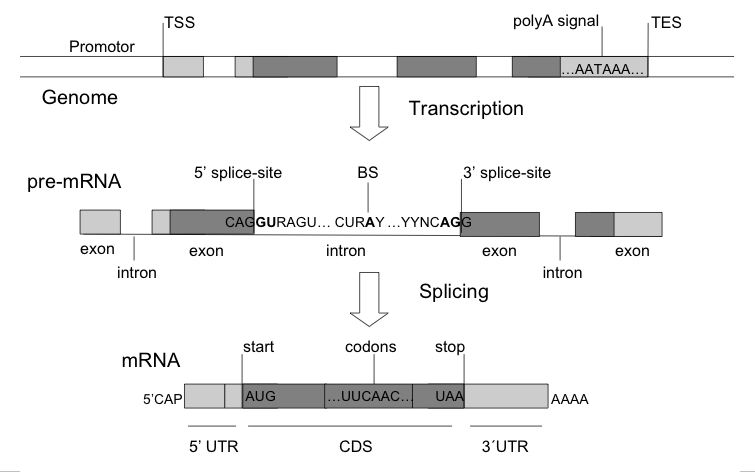
The signals are tipically the splice-sites (donor: GTRAGT, acceptor: NCAG, branch-site: CURAY),
the start of translation (ATG), and the end of translation (TGA,TAA,TAG). The content sensor most commonly
used is the bias in codon usage: coding regions use some codons more frequently than others. Both, signal sensors
and content sensors must be trained, i.e., we must start from a set of observations (know genes) from which
we build our model. Predicting a gene is therefore looking for new elements in the genomic sequence that look like our model.
This can be established in terms of probabilities.
- Comparative methods: They are also called extrinsic. They include two strategies:
those that use homologies with sequences from other genes, also called homology-based, and those that make
comparisons with genomic sequence from other genomes, also called comparative.
- Homology-based: these methods predict a gene using the alignment of a protein
(or RNA sequence in the form of full-length mRNA, cDNA or EST) with the genome
sequence that we want to annotate. The known sequence (also called evidence)
guides the prediction. There are several ways to
achieve this: the simplest way consist in accepting the alignment of the known sequence to the genome as
the gene prediction. More sophisticated methods use the known sequence as a guide and try to complete the evidence into
a complete gene structure. The efficacy of this method depends on the number of known gene sequences, hence it is
limited by how complete the biolodical databases are.
- Comparative-genomics-based: These methods are based in the hypothesis that the
sequences conserved between to genomes relatively close to each other are
functional, and therefore, possibly coding for a gene.
In fact, the annotation of a genome involves a combination of several methods of
gene prediction, and perhaps the prediction of other biological signals
(like transcription start sites, promoter regions, etc).
In this practical we will try different methods of the type ab-initio and homology-based.
|
|
A. Gene annotation: using a genome annotation browser
|
Step 1. Identification of a known gene
- Open the sequence for this genomic sequence in a different window: FASTA SEQUENCE.
- Open the UCSC Genome Browser
- Select the BLAT (human) link to locate
the genomic coordinates of our anonymous sequence
- Paste the DNA sequence
- Submit the file
- Click over the first hit: (browser link)
- In this picture, can you find your sequence? Which gene is this one?
- Try to understand the following options (use Refresh):
- RefSeq Genes
- Vega Genes
- SNPs
- Conservation
- Use the Zoom in and out (specially to find the neighbour genes)
 |
 |
|
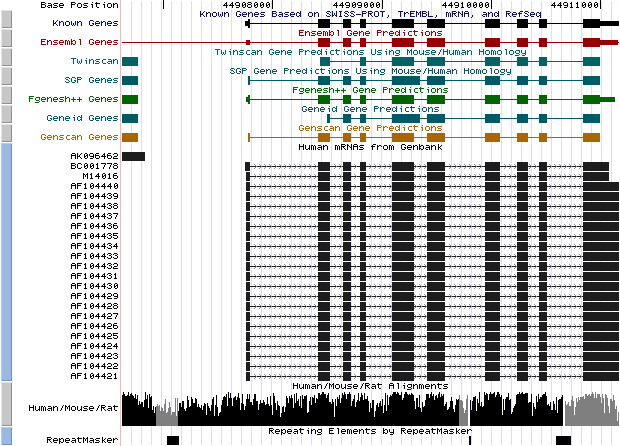
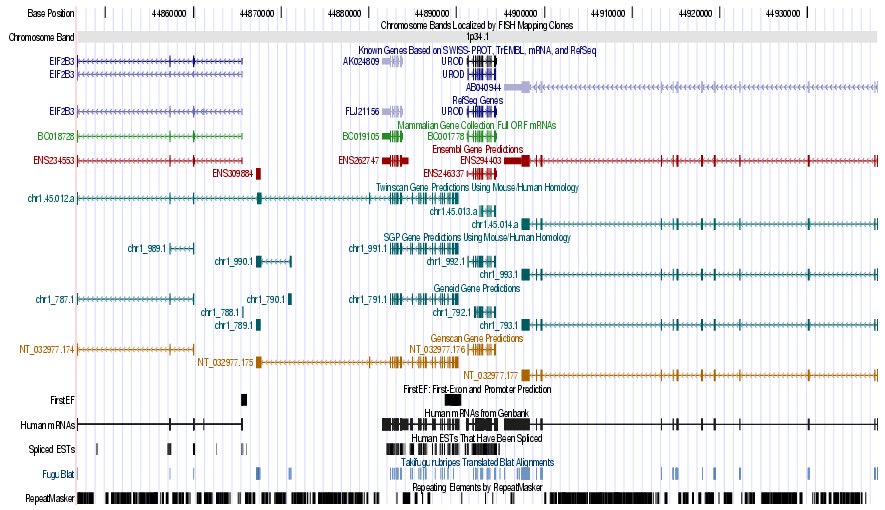
Figure 7. (a) UCSC genome browser representation of the region
containing the gene uroporphyrinogen decarboxylase (URO-D) (b)
UCSC genome browser representation of the contex (100Kbps) region around
the gene uroporphyrinogen decarboxylase (URO-D).
|
|
Step 2. The coding annotation of the URO-D gene:
The FASTA SEQUENCE used corresponds to (a version of) the
genomic region containing the URO-D gene.
Defects in UROD are the cause of porphyrya, a group of rare,
inherited blood disorders in which cells fail to change chemicals (porphyrins)
to the substance (heme) that gives blood its color.
|
|
B. Exploring ab initio gene prediction
|
Step 3. Running geneid
- Connect to the geneid server
- Paste the FASTA sequence of the genomic region of the URO-D gene
- Choose geneid output format
- Run geneid with different parameters:
- Searching signals: Select acceptors, donors, start and stop codons. Look for them in the real annotation of the sequence
- Searching exons: Select All exons and try to find the real ones
- Finding genes: You do not need to select any option (default behaviour).
Compare the predicted gene with the real gene, with CDS coordinates:
1107..1126
1748..1860
1976..2055
2132..2194
2434..2631
2749..2910
3279..3416
3576..3676
3780..3846
4179..4340
 |
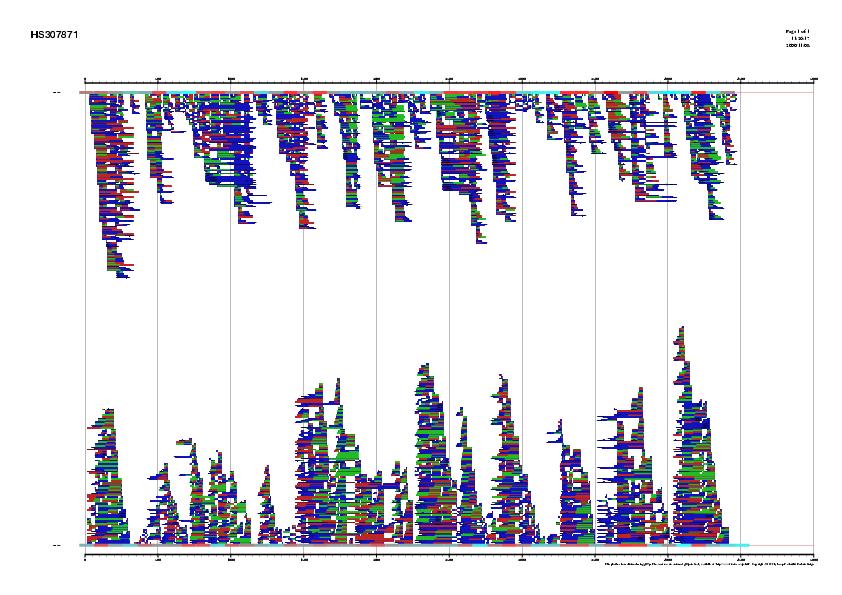 |
 |
|
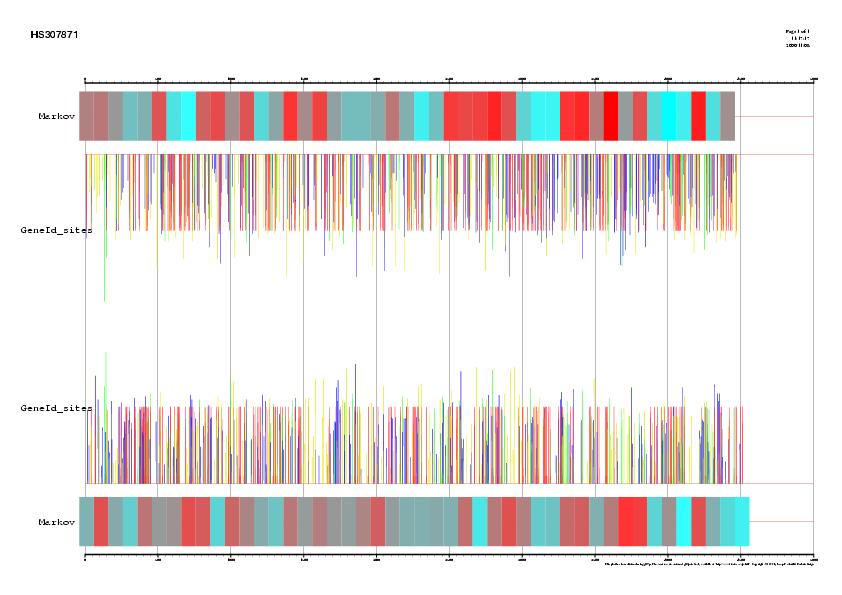
Figure 1. Signal, exons and genes predicted by geneid in the
sequence HS307871
|
|
Step 4. Running other genefinders
Provided that there are several alternative programs to analyze a DNA
sequence, we can run every application and observe the common parts of the
predictions.
- GENSCAN:
- Connect to the GENSCAN server
- Paste DNA sequence
- Press Run Genscan
button
Compare annotations and predictions
FGENESH:
- Connect to Softberry homepage
- On the left frame, select GENE FINDING in Eukaryota
- Select the program FGENESH
- Paste DNA sequence
- Press Search button
- Compare annotations and predictions
GRAIL:
- Connect to GrailEXP homepage
- Activate Perceval Exon Candidates box
- Paste DNA sequence
- Press Go! button
- Check the results
- Compare annotations and predicted exons
NOTE: First exon is always missed in the predictions and there are some
problems to detect the donor site from exon 5. Detection of Start codons is
a serious drawback in current gene finding programs (see Figure 2). However,
this problem can be overcome by using homology information to complete the
gene prediction.
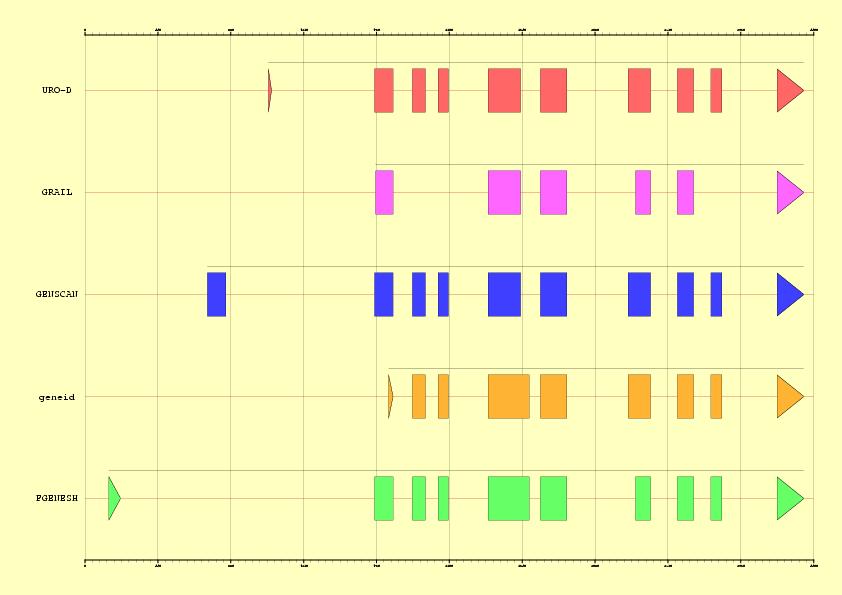
|
Figure 2. EMBL annotation and genes predicted by Grail,
GENSCAN, geneid and FGENESH in the sequence HS307871
|
|
|
C. Using EST/cDNA homology information
|
Step 5. Using GrailEXP
- Connect to GrailExp homepage
- Activate Galahad EST/mRNA/cDNA Alignments box
- Select GrailEXP database (RefSeq/HTDB/dbEST/EGAD/Riken)
- Activate exon assembly: Gawain Gene Models
- Paste DNA sequence
- Press Go!
button
Check the results: predictions and supporting information
Compare annotations, ab initio GRAIL prediction and five predicted
alternative spliced variants
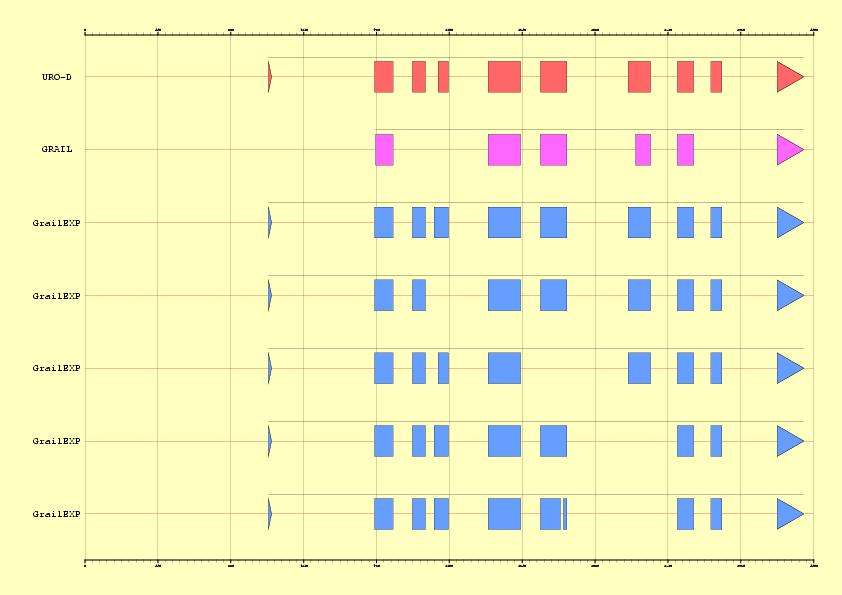
|
Figure 3. Comparison between EMBL annotation and genes predicted ab inition by Grail Vs five alternative predictions supported by ESTs information
in the sequence HS307871
|
Step 6. Using other gene finding programs + alignment of transcripts
Using blastn, we can search
the database est_human for ESTs supporting
future predictions. Filter this output in order to select those
non-overlapping ESTs that could form a complete cDNA sequence (see Figure 4).
Moreover, ESTs not divided into two or more pieces in the genomic sequence
(containing a couple of splice sites) should be rejected.
- Connect to the FGENESH-C server
(on Gene finding with similarity menu)
- Paste the sequence HS307871
- Paste the cDNA sequence or EST you have selected
- Press the search button
- Notice that the predicted gene will be necessarily supported by homology
information, so the prediction will most likely only cover in the genomic region overlapping
your EST query.
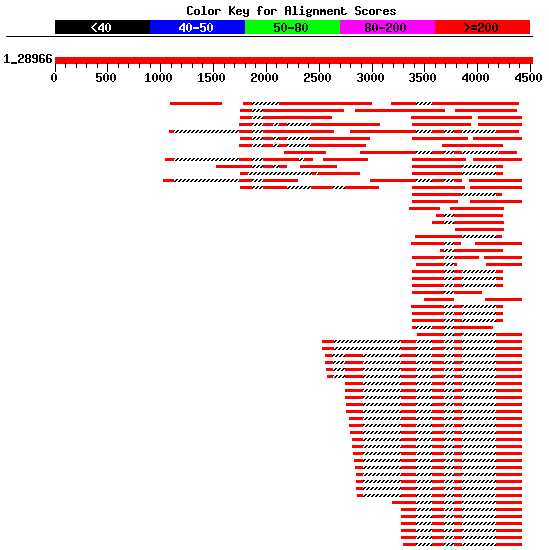 |
|
Figure 4. Best human ESTs in the alignment mapped on the genomic sequence HS307871
|
|
|
D. Using protein homology information
|
Step 7. Spliced alignment
A tool capable to produce Spliced alignments is very useful in the
homology-based prediction. For instance, if we have
a homologous protein sequence.
In this case, gene prediction is carried by producing an alignment of the protein sequence to the genome, and
at the same time, fitting the alignment to the best splice sites predicted in the genomic sequence.
- Open the NCBI blast server
- Choose blastx program (genomic query versus protein database)
- Paste the genomic sequence and press the
Blast! and
Format!
- Select the first protein. Display the FASTA sequence or click
here. Obviously, it is the known protein URO-D, which is already
annotated in the genomic sequence (see UCSC browser).
- Open the GeneWise web server
to use this protein to predict the best gene structure
- Paste both protein and genomic sequences and run the program
- Compare the predicted gene and the EMBL annotations (above). Look at the splice-sites
given by GeneWise to check whether the exon boundaries are correct.
 |
Figure 5. Best HSPs representing proteins homologues similar to the
genomic sequence HS307871 obtained using blastx
|
|
Step 8. Spliced alignment using homologous proteins
From the blastx output, choose several homologous genes from other species (different from human) and run genewise for each
one separately, again. Observe the loss of accuracy as the homologue is further from the original human protein:
|
Step 9. Using protein homology information: GenomeScan
Protein homology information can also be used to enhance ab initio predicted
exons supported by blastx HSPs as in the case of GenomeScan and
geneid improving therefore the final prediction
GenomeScan:
- Connect to the
GenomeScan web server
- Retrieve the protein from the previous blast search
- Paste both genomic and protein sequences
- Press the button GenomeScan
- Check the results. It seems that the first exon has not been detected
even using homology information. This is due to the fact that blast programs
have a minimal word lenght.
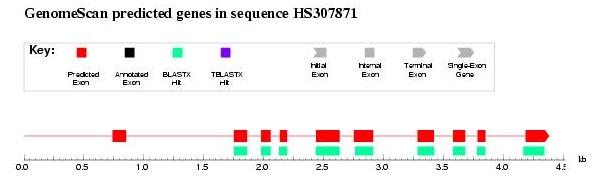 |
|
Figure 6. GenomeScan output: first exon is not correctly predicted probably due to blast length restrictions
|
|
|
|
E. Results
|
Here you can find the solutions to every exercise:
|
|
|
F. Bibliography
|
- J.F. Abril and R. Guigó.
gff2ps: visualizing genomic annotations. Bioinformatics 16:743-744 (2000).
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W. & Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 215:403-410 (1990).
- Burge, C. and Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78-94 (1997).
- E. Blanco, G. Parra and R. Guigó.
Using geneid to Identify Genes. In A. D. Baxevanis and D. B. Davison,
chief editors: Current Protocols in Bioinformatics. Volume 1, Unit 4.3.
John Wiley & Sons Inc., New York. ISBN: 0-471-25093-7 (2002).
- G. Parra, E. Blanco, and R. Guigó.
Geneid in Drosophila. Genome Research 10:511-515 (2000).
- Asaf A. Salamov and Victor V. Solovyev. Ab initio Gene Finding in
Drosophila Genomic DNA Genome Res. 10: 516-522 (2000).
- Yeh, R.-F., Lim, L. P. and Burge, C. B. Computational inference of
homologous gene structures in the human genome. Genome Res. 11: 803-816 (2001).
-
D. Hyatt, J. Snoddy, D. Schmoyer, G. Chen, K. Fischer, M. Parang, I. Vokler, S. Petrov, P. Locascio, V. Olman, Miriam Land, M. Shah, and E. Uberbacher.
Improved Analysis and Annotation Tools for Whole-Genome Computational Annotation and Analysis: GRAIL-EXP Genome Analysis Toolkit and Related Analysis Tools.
Genome Sequencing & Biology Meeting (2000).
- Ewan Birney and Richard Durbin. Using GeneWise in the Drosophila
Annotation Experiment. Genome Res. 10: 547-548 (2000).
|
|
