The major decision was choosing the organism we were going to use as a reference to search for conserved selenoproteins and its machinery proteins in the Colinus virginianus’ genome. After some searching in the SelenoDB 2.0 database, we decided to use the chicken (Gallus gallus), because it was a bird and therefore more closely related to C.virginianus than the other organisms. It is important to mention that we had the chance to use the turkey (Meleagris gallopavo), but we decided to choose the chicken because we consider that it is a better studied organism than the others, so it should be better characterised.
From this database we obtained the FASTA sequences that served as queries to compare with the C.virginianus’ genome. We pasted each sequence in a different EMACS file that was named as Query.fa, where Query refers to the protein name.
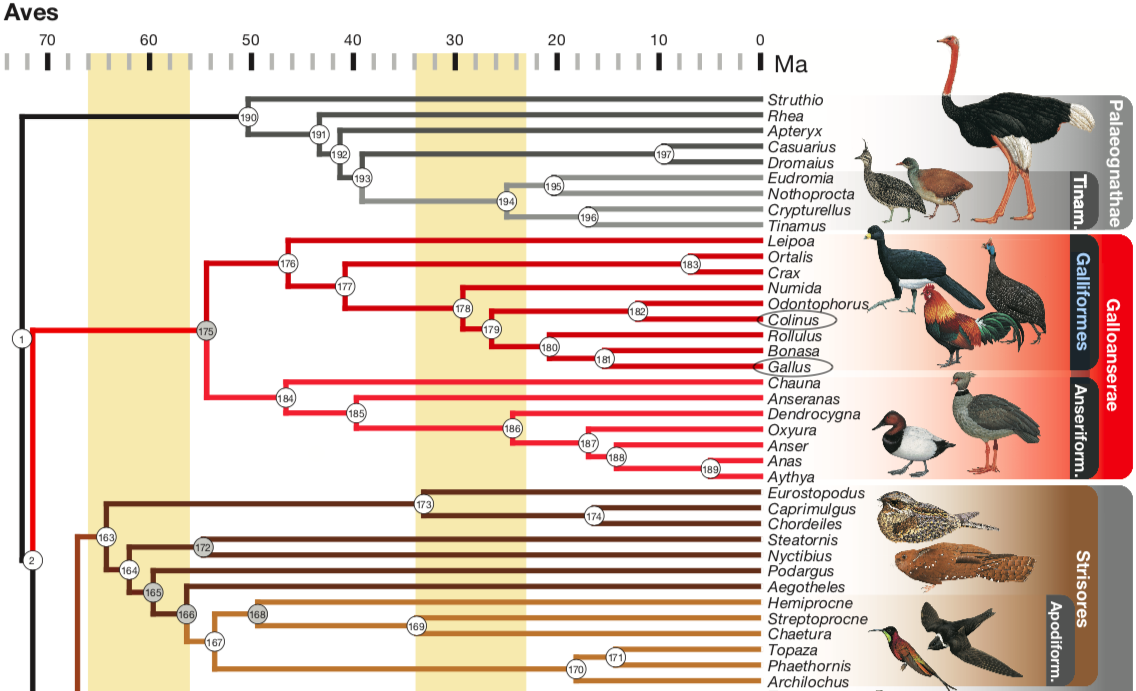
Figure 5: Fragment of Phylogeny of some birds. As we can see, both Collinus and Gallus genera belong to the order of Galliformes, having differed less than 30 M years (a not specially long time). This fact was crucial for us to decide using Gallus gallus as the reference animal. Completed phylogenetic tree can be found in Prum et al,. 2015 (see References).
At this point, we wanted to develop a program to analyse the possible selenoproteins (and machinery proteins) faster. With this program we were able to perform all the steps necessary for the analysis automatically except for:
1-. The introduction of each protein’s name, which was introduced manually by us at the beginning of the program.
2-. The scaffold selection, which was chosen manually by us after each tblastn.
3-. The first and last nucleotide position, that were chosen manually by us before each fastasubseq.
You can download our program by clicking here.
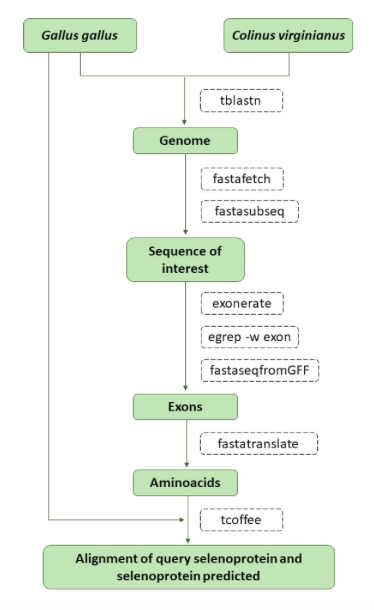
Before performing the tblastn program, we ought to change the U that represents the selenocystein to an X (in case there was any). That way the tblastn program could read the full sequence without any error. This action is accomplished with the following command line:
sed s/U/X/g $prot > $prot.x
Once we had changed the possible U to X, we were ready to perform the tblastn program. This program makes a comparison between the query protein and the C. virginianus’ genome that gives an output containing the significant hits it has found. Our program considered only those hits with an e-value equal or minor to 0.001. In order to perform the tblastn, our program contains the following command line:
tblastn -query $prot.x -db /mnt/NFS_UPF/soft/genomes/2019/Colinus_virginianus/genome.fa -out $prot.tblastn -outfmt 7 -evalue 0.001
As we mentioned before, the scaffold selection was made manually by us after each tblastn; and the criteria to do so was picking the scaffold with the lowest e-value, and from that scaffold we chose those hits with more than 70% of identity. In other words, we chose the most significant scaffold for each protein. Since the tblastn is an heuristic program, we considered that it was not necessary to pick other scaffolds, because their significance was stated lower by the program and they had less hits, which did not match our objective to achieve the highest possible significance.
However, with proteins belonging to the same family, it was difficult to distinguish which scaffold was the most suitable for each of them. In these cases, we picked those scaffolds with an e-value equal or minor to 0.001 and with the highest percentage of identity possible. Then, we performed a phylogenetic tree in order to choose the best scaffold for each protein.
The fastafetch program allowed us to extract the scaffold with the possible selenocysteines from the C. virginianus’ genome, which was done with the following command line:
fastafetch /mnt/NFS_UPF/soft/genomes/2019/Colinus_virginianus/genome.fa /mnt/NFS_UPF/soft/genomes/2019/Colinus_virginianus/genome.index $scaffold > $prot.fastafetch
As we mentioned before, the first and last nucleotide position were chosen manually by us before each fastasubseq. To do so, we ignored those that were very far away from the others (only in a few proteins), or the ones that, despite being in the same scaffold, had a very high e-value and a low percentage of identity; both situations used to happen together.
Doing this was necessary because then we avoided problems of excessive length in the subseq file when performing the Seblastian or SECISearch3, otherwise those webservers emitted an error and some subseq files could not be processed for being too large.
After defining the positions, we extended them 50,000 more nucleotides in both 5 prime and 3 prime ends in order to avoid missing part of the C. virginianus’ sequence due to the presence of introns (the scaffold only contains the coding region). But there were two special situations to take into account:
1-. If the first position had a negative number (meaning that it begun before the first nucleotide of the genome), the first nucleotide position was set to 1.
2-. If the last position had exceeded the scaffold length, the last nucleotide position was set to the last position of the scaffold.
It is important to mention also that the scaffold length was calculated by multiplying the number of lines minus one in the fastafetch file by the number of nucleotides in one row. Finally, the length we have used to do the fastasubseq was the last nucleotide position minus the first nucleotide position (after applying all changes specified above).
Once we extracted the scaffold thanks to the fastafetch program and we defined the start and length position, we were able to obtain the region of interest with the fastasubseq program using the following command line:
fastasubseq $prot.fastafetch $start $length > $prot.subseq
With exonerate we were able to generate a FASTA file containing the exons of the predicted protein, using the following command line:
exonerate -m p2g --showtargetgff -q $prot.x -t $prot.subseq | egrep -w exon > $prot.exonerate.gff
This program allowed us to obtain the cDNA from the FASTA file created with the exonerate, using the following command line:
fastaseqfromGFF.pl $prot.subseq $prot.exonerate.gff > $prot.pred.nuc
Fastatranslate transforms the cDNA obtained with the fastaseqfromgff into the protein sequence. It is important to mention that we used the first option of the program (-F 1). This was done by our program with the following command line:
fastatranslate -F 1 $prot.pred.nuc > $prot.pred.aa
fastaseqfromGFF.pl $prot.subseq $prot.exonerate.gff > $prot.pred.nuc
Before performing the t-coffee, it is necessary to change the * to an X for the correct functioning of the program, which is done with the following command line:
sed s/*/X/g $prot.pred.aa > $prot.pred.aa.x
Finally, we performed a global alignment between the query protein and the predicted protein sequence using the t-coffee program with the following command line:
t_coffee $prot.x $prot.pred.aa.x > tcoffee_$prot
Here ends our program, and the following step was performed in the Seblastian/SECISearch3 webpage.