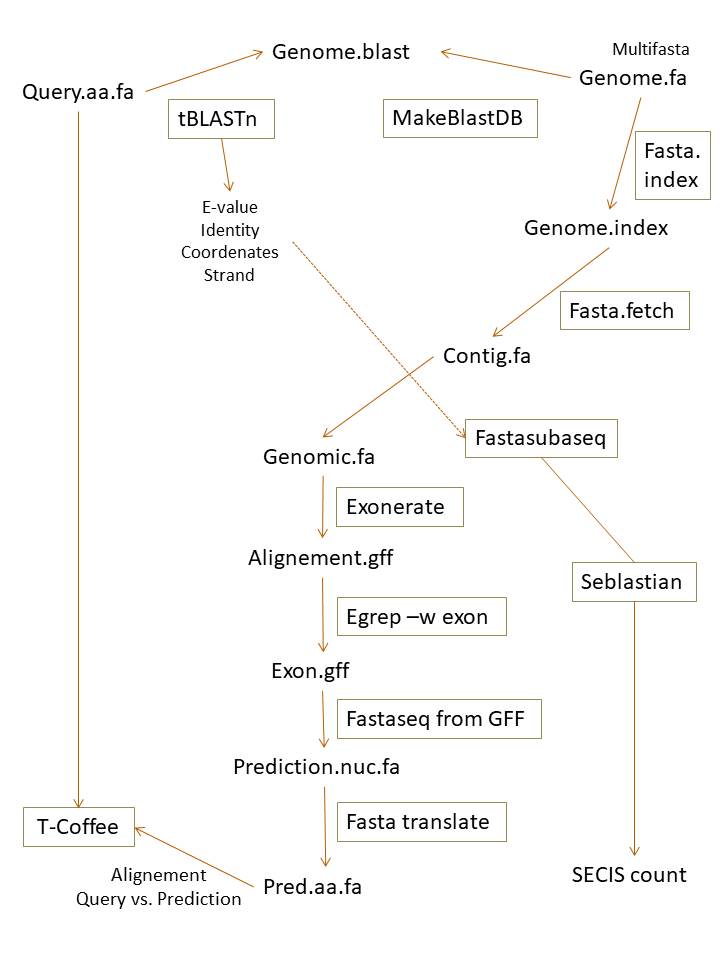
The aim of this project was to identify and annotate the selenoproteins and the machinery required for their synthesis encoded in Mus spicilegus genome. In order to do that, we selected a well anotated reference genome where selenoproteins have been described, for that we used the database SelenoDB 1.014. In our case, we selected Mus musculus because it is the phylogenetically closest specie with a very-well-annotated selenoproteome. The database SelenoDB 2.015 was also checked whether to see if there are other selenoproteins also present in Mus spicilegus. The following scheme shows all the steps:
Image 6. Procedure scheme.
The Mus spicilegus genome was obtained following this path:
/mnt/NFS_UPF/bioinfo/BI/genomes/2018/Mus_spicilegus/genome.fa
As mentioned previously, amino acid sequences of the queries of Mus musculus (selenocysteines proteins, homologous and selenoprotein machinery) were obtained from SelenoDB 1.0 database. Every sequence was copied into an EMACS file named as query.aa.fa, where “query” is the abbreviation of the protein named used on SelenoDB 1.0. However, not all the selenoproteins or machinery proteins were found in this database, so for the missing ones the database SelenoDB2 was used.
Selenocysteines are representedon the query as a "U". However, the software packages required for the analysis do not recognise this character. This is why all the “U” must be replaced by an “X”, which stands for any possible amino acid. Furthermore, all the symbols (%, #, @, etc) found at the end of the sequences, which are not recognised by the softwares, were removed.
Queries adquisition
In order to agilise the procedure, a semiautomatization was performed. We elaborated a pearl program. To explain how it works, let's go step by step:
First of all, all "U" characters must be substituted by "X" because softwares used can not detect the firt one. The file obtained is named as query.fa, were “query” is the abbreviation of the protein named used on SelenoDB 1.0. Once done, it appears a message on the screen that indicates that U have been changed.
Then, Tblastn was performed in order to translate Mus spicilegus genome to amino acids and align it with Mus musculus query. Here we obtain different scaffolds and we have to choose which is the more complete one that has a better e-value, which is the probability to obtain a random result. The lower the e-value, or the closest it is to zero, the more significant the match is. The file obtained is called as query.blast.fa.
The program asks you to say which is the name of the scaffold you want to extract, you select this information from the blast manually and introduce it. The file obtained is named query.fastafetch.fa. A message appears on the screen to inform that this process has finished.
Here, the program starts by calculating the lenght of the scaffold you introduced and print it on the screen. Then, it asks you to introduce the start and final position as well as the lenght of the scaffold used.You catch all this information from the blast manually and introduce it except the lenght that you have just to copy and paste it from the screen. The file obtained is named query.fastasubseq.fa. A message appears on the screen to inform that this process has finished.
On this step, the program translates the Mus musculus query to nucleotides and compares it with the sub-sequence of the scaffold selected. Furthermore, with "grep" command, we extract the exons of this regions and redirect them to another file named query.exon.gff. A message appears on the screen to inform that this process has finished.
Then, the program translates concatenate the exon sequences of Mus spicilegus extracted as a single one on a file named query.nt.fa. A message appears on the screen to inform that this process has finished.
Next step consists on translating the query.nt.fa to amino acids on a file named query.pep.fa. A message appears on the screen to inform that this process has finished.
Finally, the alignment of query.pep.fa (Mus spicilegus) and query.fa (Mus musculus) is performed and we obtain a file named query.tcoffee.txt. A message appears on the screen to inform that this process has finished.
In order to verify if the predictions made were selenoproteins, we used the program SECISearch/Seblastian18 to predict SECIS elements in the corresponding sequence.
We introduced in the program the sequence obtained in the Fastasubseq output file.