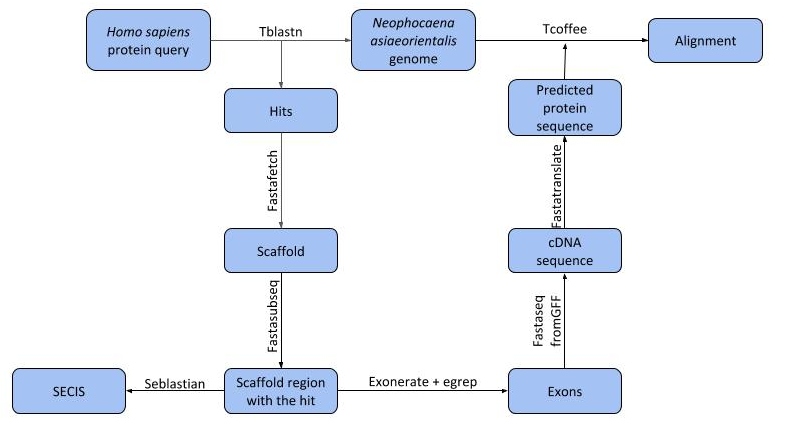
1. Obtention of the genome
The genome of Neophocaena asiaeorientalis was provided by the professors of bioinformatics. We downloaded it from the file:
/mnt/NFS_UPF/bioinfo/BI/genomes/2018/Neophocaena_asiaeorientalis/genome.fa
Also, an indexed genome was provided, which was obtained from:
/mnt/NFS_UPF/bioinfo/BI/genomes/2018/Neophocaena_asiaeorientalis/genome.index
We observed that the genome is composed by 128596 scaffols, which means that it is hugely fragmentated.
2. Obtention of the queries
The genome of reference that was used is Homo sapiens genome, as it is the closest species to the one of study (Neophocaena asiaeorientalis) with a well-annotated genome. Homo sapiens proteins were extracted, in most cases, from SelenoDB1[10], a database checked manually. In some of them, we had to extract their sequence from the SelenoDB2 because they weren't in the other database. A file with the Homo sapiens proteome was downloaded and each protein was coppied into a separated file, named prot.fa.
In some proteins, the predictions were not good enough, so we also compared our species of study with Tursiops truncatus, a model which is worse annotated, but phylogenetically closer. The selenoproteome of Tursiops truncatus was obtained from SelenoDB2, as it was not present in the SelenoDB1.
3. Substitution of U to X
The sequences that contained a selenocysteine were modified, changing the "U" aminoacid for an "X". That was done because some of the bioinformatic resources used do not recognise the letter "U" as an aminoacid, and this would have lead to errors. Other characthers as "#", "@" and "*" were also removed to avoid errors.
4. Prediction of selenoproteins
Scaffold search: tblastn
BLAST (Basic Local Alignment Search Tool) is a program that compares biological sequences to find regions of similarity [11]. This program is based in an heuristic algorism, which uses the most efficient alignment strategy in order to find the most realistic results.
The aim of this study is to find regions of our genome (Neophocaena asiaeorientalis) that are highly likely to encode a selenoprotein. With this objective, first we compared the genome of our species with the different querys that we obtained from Homo sapiens and Tursiops truncatus.
To do so, the specific program TBLASTN was used, which allows to find regions of similarity between sequences. It compares translated genomic sequences vs proteins.
The option "-outfmt 6" was used, in order to obtain the result of the blast tabulated.
The command used to execute this step was the following:
tblastn -query prot.fa -db genome.fa -out prothuman.blast -outfmt 6
Obtention of the best scaffold candidates
After performing TBLASTN, an output was obtained with all the potential matches between the queries and the genome of Neophocaena asiaeorientalis. The best scaffold candidate was selected from the list obtained, taking into account different parameters: the length, a high score, and a low e-value.
Obtention of the exons
The following step consists on extracting those parts of the genome of Neophocaena asiaeorientalis that match with the protein of the Homo sapiens or Tursiops truncatus genome. For this purpose we used Exonerate. Before executing this program, we run two programs. First, the program fastafetch was used to generate a file with the scaffold sequence and then, the program fastasubseq was used to extract from the scaffold the region of interest.
a) Fastafetch
The fastaindex file was already provided by the professors. The command used to execute this step was the following:
fastafetch genome.fa genome.index $scaffold > $scaffold/$proteina.$scaffold.fastafetch.fa
b) Fastasubseq
Before running fastasubseq, we extended the region selected 50000 nucleotides on each side to make sure that the whole gene sequence was extracted. Besides, to reinforce the idea that the limits chosen were inside the scaffold, we used two comands. The first one, to make sure that the lower limit was not less than 0, and the second one, to make sure that the upper limit does not exceed the length of the scaffold.
The fastasubseq was executed using the following command:
fastasubseq $proteina.$scaffold/$proteina.$scaffold.fastafetch.fa $start $lenght > $proteina.$scaffold/$proteina.$scaffold.fastasubseq.fa
c) Exon extraction
After selecting the subsequence, Exonerate was used to compare the genome of reference with the query. With this process, we obtained the exons of the extracted region.
The command used to execute this step was the following:
exonerate -m p2g --showtargetgff -q /home/u124036/grup10/$proteina.fa -t $proteina.$scaffold/$proteina.$scaffold.fastasubseq.fa | egrep -w exon > $proteina.$scaffold/$proteina.$scaffold.exon.gff
Obtention of the cDNA
With the file that contains the exons, a new file was created that corresponds to the cDNA of the extraction.
The command used to do so is the following:
fastaseqfromGFF.pl $proteina.$scaffold/$proteina.$scaffold.fastasubseq.fa $proteina.$scaffold/$proteina.$scaffold.exon.gff > $proteina.$scaffold/$proteina.$scaffold.pred.nt.fa
Obtention of the Selenoprotein
The following step was to translate the cDNA sequence into protein. For that purpose, we used the fastatranslate command.
The command was the following:
fastatranslate -f $proteina.$scaffold/$proteina.$scaffold.pred.nt.fa -F 1> $proteina.$scaffold/$proteina.$scaffold.pred.aa.fa
Substitution of "*" to "X"
To avoid mistakes when running tcoffee, we changed all the "*" present in the translated protein to "X" with the command sed 's/*/X/g'.
Comparing our alignment result with the protein problem: T-coffee
We used T-coffee (Tree-based Consistency Objective Function for alignment evaluation) for obtaining the alignment between the Homo sapiens or Tursiops truncatus query and the predicted protein of Neophocaena asiaeorientalis[12]. The command used to perform T-coffee was the following:
t_coffee $proteina.$scaffold/$proteina.fa $proteina.$scaffold/$proteina.$scaffold.predX.aa.fa > $proteina.$scaffold/$proteina.$scaffold.tcoffe.txt
5. Confirmation of Exonerate prediction: Genewise
Genewise software was used to validate the Exonerate prediction obtained [13].
The command used was the following:
Genewise -pep -pretty -cdna -gff -trev query protein.genomic.fa > protein.Genewise
With the results obtained from the Genewise, another t-coffee aligment was performed to finally compare the two different outputs obtained (Exonerate and Genewise).
6. Seblastian search
Seblastian software was used to predict the SECIS found in each protein. We only took into account the SECIS present in the 3'-UTR region of the same strand of the protein. In some cases where Seblastian did not find any selenoprotein, we used the SECISearch3 to obtain more information about the SECIS [14]. The input was the file obtained in the fastasubseq step.
Figure 6. Scheme with all the steps followed in order to predict the selenoproteome and the machinery proteins of Neophocaena asiaeorientalis.
7. Automation
In order to make the process easier and faster a program was made to automatize some of the steps.
It starts after selecting manually the best scaffolds of the tblastn output.
The program can be downloaded here.
8. Phylogenetic tree obtention
We created a phylogenetic tree with all the proteins analysed in order to see if the predicted proteins of Neophocaena asiaeorientalis were close to the initial queries of Homo sapiens. This tree was created with Phylogeny.fr