In this project, we aim to predict and annotate all the selenoproteins present in Helostoma temminkii, as well as the machinary and the proteins related to the metabolism of selenium. In order to do that, we compared our species with another fish, Danio rerio, most commonly known as Zebrafish, due to their close phylogenetic relationship and to the fact that selenoproteins from Zebrafish are annotated and described. The analysis followed the scheme shown below:
The genome of Helostoma temminkii (genome.fa) was downloaded from the file provided by the bioinformatics teachers present in the following directory:
/mnt/NFS_UPF/bioinfo/BI/genomes/2018/Helostoma_temminkii/genome.fa
We also obtained the indexed genome from:
/mnt/NFS_UPF/bioinfo/BI/genomes/2018/Helostoma_temminkii/genome.index
As previously mentioned, the Danio rerio genome was chosen to identify the selenoproteins in Helostoma temminkii.
The sequences of the queries were obtained from SelenoDB 1.0 database.
Once we copied the sequence, we pasted it into an EMACS file named as query.fa, where Query is the protein’s name. Generally, the name is the same as the one used in the database, but in some cases we called the proteins query_1, query_2... when those proteins were cataloged as “NONE”.
In order to correctly perform the analysis, we changed the character “U” for an “X”, which represents any possible amino acid, due to the fact that the softwares didn’t recognise the “U” as one. Also, we eliminated the symbols, like #, that were at the end of the sequences.
In order to explore the potential selenoproteins faster, an automatic perl program was developed. The analysis of the files obtained and final predictions were performed afterwards by us.
This program performed all the needed steps except for query acquisition and the SECIS prediction. Also, we developed another perl program without the Blast alignment, in order to double check that the queries were selected correctly.
Both programs required us to manually introduce the name of the protein, the name of the scaffold, the first nucleotide and the last nucleotide aligned and the length of the scaffold.
Blast
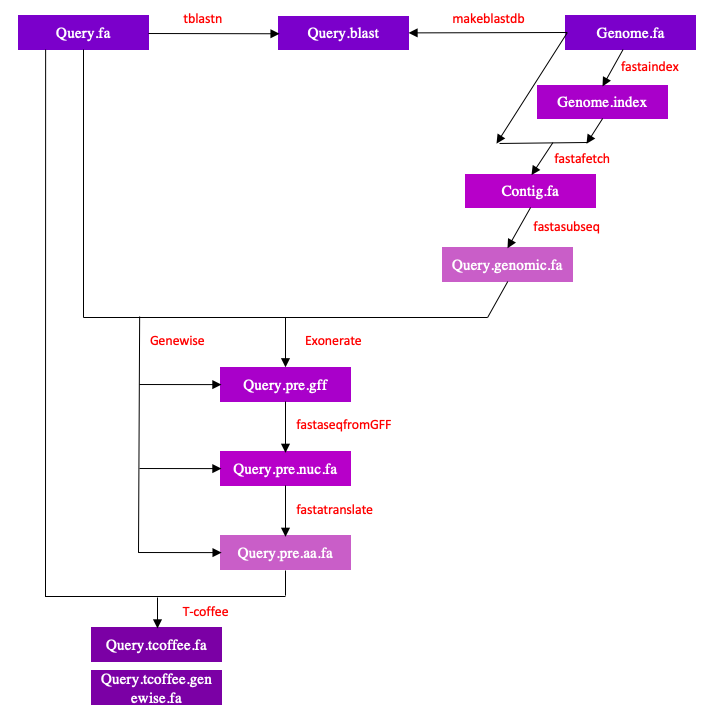
Tblastn compares the protein query to the Helostoma temminkii’s genome, in order to be able to choose the scaffolds with significant hits. Our program that had Blast analysis automatized selected the scaffold if the hits had an E-value equal or minor to 0.01. We manually followed the same criteria. The output is directed to the $p.blast document, and it contains all the information.
tblastn -query $p.fa -db /mnt/NFS_UPF/bioinfo/BI/genomes/2018/Helostoma_temminkii/genome.fa -out $p.blast -outfmt 6 -evalue 0.01
Fasta fetch
With this command we are able to extract the scaffold from the hits of the Helostoma temminkii’s genome.
fastafetch /mnt/NFS_UPF/bioinfo/BI/genomes/2018/Helostoma_temminkii/genome.fa /mnt/NFS_UPF/bioinfo/BI/genomes/2018/Helostoma_temminkii/genome.index '$s' > $s.fa
Fasta subseq
Once we have extracted the scaffold, we proceed to obtain the region of interest. In order to avoid missing part of the sequence because of the presence of introns (the hit only correspond to the coding sequences), we extended the search until 50.000 more nucleotides in both directions (5’ and 3’). This step was done automatically, and when the 50.000 exceed the sequence length, the program reduced 1 nucleotide at the time until it reached the maximum length.
fastasubseq $s.fa $start $length > $p.genomic.fa
Exonerate
This command allows us to generate a fasta file with the cDNA of the predicted protein from the gff file obtained with Exonerate.
exonerate -m p2g --showtargetgff -q $p.fa -t $p.genomic.fa | egrep -w exon > $p.pre.gff
Fasta seq from gff
This command allows us to generate a fasta file with the cDNA of the predicted protein from the gff file obtained with Exonerate.
fastaseqfromGFF.pl $p.genomic.fa $p.pre.gff > $p.pre.nuc.fa
Fasta translate
Fastatranslate allows us to translate the cDNA into the protein sequence.
fastatranslate $p.pre.nuc.fa -F 1 > $p.pre.aa.fa
T-COFFEE
T-coffee allows us to perform a global alignment between the predicted protein sequence and the query protein from the reference species, Danio rerio.
t_coffee $p.fa $p.pre.aa.fa > $p.tcoffee.fa
Genewise
At this point, the program asks us if the gene is located in the positive or the negative strand (+ or -). If it is the positive the commands are the ones below:
genewise -pep -pretty -cdna -gff $p.fa $p.genomic.fa > $p.genewise.info.txt
genewise -pep $p.fa $p.genomic.fa > $p.genewise.raw.fa
If it is the negative strand the program does the commands written below:
genewise -pep -pretty -cdna -trev -gff $p.fa $p.genomic.fa > $p.genewise.info.txt
genewise -pep -trev $p.fa $p.genomic.fa > $p.genewise.raw.fa
Then, from these files the program selects the following ones:
grep -v -e 'Making' -e '//' $p.genewise.raw.fa > $p.genewise.fa
And finally, the T-COFFEE is executed with the Genewise prediction in order to compare this T-coffee output with the T-coffee obtained using the Exonerate software.
t_coffee $p.fa $p.genewise.fa > $p.tcoffee.genewise.fa
SECIS, SEBLASTIAN and/or SECISearch3
As previously explained in the Introduction, SECIS are necessary elements in selenoporteins mRNA in order to present Sec in their sequences.
We use Seblastian in order to identify SECIS elements in our predicted sequences. If no SECIS elements were predicted, we also used SECISearch3 in order to corroborate these findings.
In the case of finding more than one possible SECIS, we established as a criteria that the SECIS structure must be on the 3’-UTR of the gene and it has to be on the same strand as the coding gene of the selenoprotein.
Phylogenetic tree
Once we have all the proteins, we make the phylogenetic trees for several protein families using the online tool Phylogeny.fr. The output file provides the correlation between the reference species query and the predicted protein in our species.