The main aim of this research was to identify all the selenoproteins and selenoprotein machinery genes from Helostoma temminkii. Danio rerio’s (commonly known as Zebrafish) was used as a reference species in order to find these proteins based on homology. We chose Danio rerio’s genome since it has the most well characterized and studied fish genome and its selenoproteins are thoroughly annotated. Nevertheless, Oryzias latipes (commonly known as Medaka) was also used in some situations in order to better align specific genomic locations from Helostoma temminkii which were not optimally aligned with Danio rerio’s genome and, consequently, our protein predictions were not optimal.
It is important to highlight that Helostoma temminkii, together with all the other Osteichthyes (Bony fishes) species, experienced a whole-genome duplication event named as Ts3R. As a result of this event, this subclass of fishes contain in their genome duplicated segments, as can be proved by our results for Helostoma temminkii.
For all the identified proteins in Helostoma temminkii, the analysis and discussion of the results are performed individually, emphasizing the comparison of the T-coffee outputs from Exonerate and Genewise protein predictions. We also commented on the predicted SECIS obtained using SEBLASTIAN or SECISearc3.
Finally, an schematic representation of the gene corresponding to each protein is presented and, in some cases, a phylogenetic tree is generated in order to have a better understanding of the evolutionary relations between Helostoma temminkii and other species.
The thyroid hormone deiodinases are three paralogous proteins in mammals (DIO1, DIO2 and DIO3). All three proteins are involved in reductive deodination which is essential for the regulation of thyroid hormone activity, as explained previously in the introduction. In this case, Helostoma temminkii presents a duplication for DIO3, which is also expected to be duplicated in all Bony fishes as a product of their whole-genome duplication.
When aligning each protein from the DIO family to Helostoma temminkii’s genome, four different scaffolds showed significant hits and this is due to the fact that Iodothyronine deiodinases are very similar to each other, thus, the reference protein can align to the other genomic regions also coding for DIO proteins. In order to avoid considering that a protein has been duplicated when it actually corresponds to another protein from the same family, a detailed study has been carried out in order to select the hits with the highest scores and the lowest E-values for each protein avoiding the scaffolds that were selected for the others.
DIO1
In the case of DIO1, 4 scaffolds showed significant hits when aligning with the DIO1 protein from Danio rerio (SPP00000611_2.0). However, the one that showed a lower E-value and a higher score was the one selected, OMLM01002656.1, where a selenocysteine residue can be found.
The same protein predictions were obtained with both Exonerate and Genewise, as can be seen with the T-coffee alignment. Using Exonerate, 3 exons are predicted.
The gene predicted is located between 26008-27162 in the negative strand, therefore the protein shows the same length as the one from the reference species. Nevertheless, the reference protein doesn’t include a Methionine (Met) at the beginning of the protein.
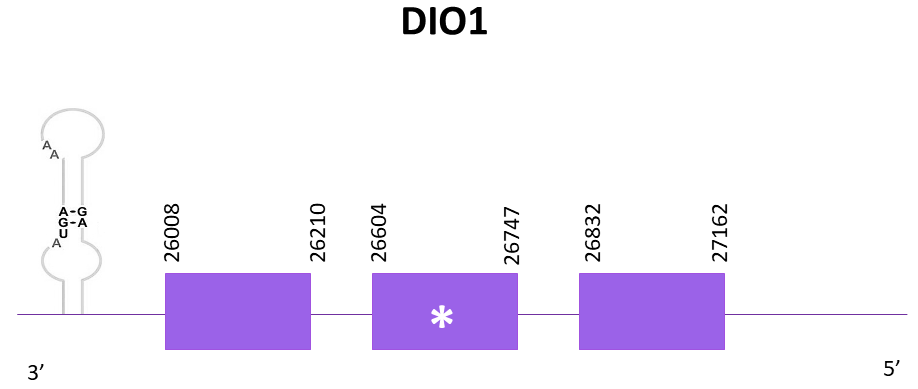
When using Seblastian, a better prediction is obtained due to the fact that the Met is included as well as a few amino acids more from the N-terminal. Two SECIS candidates are predicted at the 3'-UTR end using Seblastian.
DIO2
Four different scaffolds showed significant hits when aligning the corresponding protein from Zebrafish (SPP00000614_2.0) to Helostoma temminkii’s genome. OMLM01007644.1 was the best candidate since it had the lowest E-value and highest score.
The gene coding for DIO2 is located between 54912 and 58034 positions from the scaffold in its negative strand. 3 exons are predicted for this protein using Exonerate and a conserved selenocysteine is detected in the last exon. A SECIS structure is predicted in the 3'-UTR region.
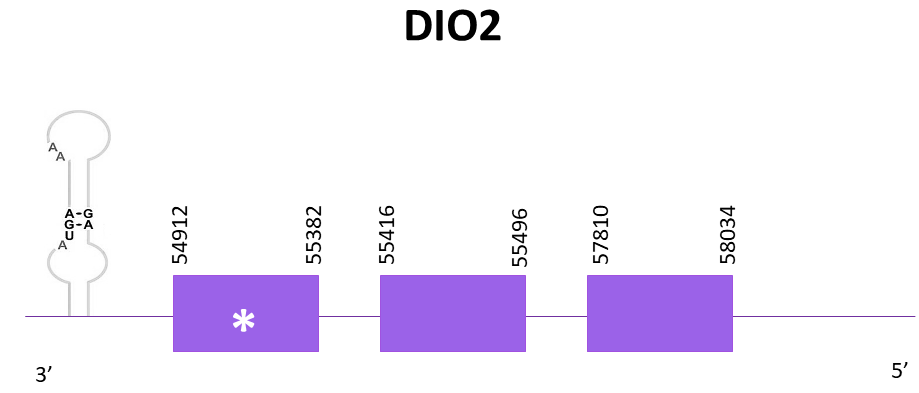
Proteins predicted using Exonerate and Genewise are pretty similar, although the one obtained with Genewise showed some gaps in its T-coffee alignment with the reference protein, whereas the one from Exonerate did not. Both predictions include the initial methionine and preserve the same amino acids in its C-terminal region as in the reference protein. Finally, using Seblastian a prediction of the protein for Helostoma temminkii is also obtained and this one appears to be identical to the one from Genewise.
DIO3a
DIO3 had 4 scaffolds showing significant hits when aligning with the DIO3 protein from Danio rerio (SPP00000612_2.0). However, the one that showed a lower E-value and a higher score was the one selected, OMLM01009181.1, where a selenocysteine residue could be found, as expected when comparing to DIO3a from Zebrafish.
The same protein predictions are obtained with both Exonerate and Genewise, as can be seen with the T-coffee alignment. Using Exonerate, 1 exon is predicted.
The gene predicted is located between 18508-19299 in the negative strand, therefore the protein shows the same length as the one from the reference species. Nevertheless, the reference protein does not include a Methionine (Met) at the beginning of the protein.

When using Seblastian, it seems that a better prediction is obtained due to the fact that the Met is included as well as a few amino acids at the C-terminal region. One SECIS element is predicted at the 3'-UTR end using Seblastian.
DIO3b
As the other DIOs, 4 scaffolds showed significant hits when aligning the corresponding protein from Zebrafish (SPP00000613_2.0) to Helostoma temminkii’s genome. However, the one that showed the lowest E-value and the highest score was the one selected, OMLM01012438.1, where a selenocysteine residue can be found, as expected.
The protein shows a worse T-coffee alignment with the reference protein when using Genewise, due to the presence of gaps, whereas the one from Exonerate did not.
The gene predicted is located between 77918-78655 in the positive strand. Even if the length of the protein is the same as the reference, both don’t include the Met and the final amino acid.

Finally, using Seblastian, the prediction obtained includes the Met as well as a few amino acids of the N-terminal, including the final stop codon, so it is a better prediction. At the 3'-UTR region one SECIS element is predicted.
To conclude, in order to check whether the predicted DIO proteins are similar to their homologues in Zebrafish we have done a phylogenetic tree that is shown below:
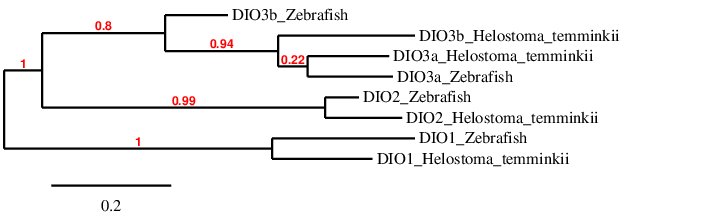
According to the phylogenetic tree, the scaffold selected for Helostoma temminkii’s DIO1 is very similar to the Zebrafish homolog, the same applies to DIO2. The predictions for the duplicated genes (DIO3a and DIO3b), DIO3b from Helostoma temminkii is more closely related to DIO3a (from both Zebrafish and Helostoma temminkii), than to the DIO3b from Zebrafish. All the predicted proteins have selenocysteine residues and SECIS predicted in the 3'-UTR end.
As previously explained in the introduction, GPx selenoproteins are involved in physiological functions and are responsible of part of hydrogen peroxide signaling, detoxification of hydroperoxides and of the maintenance of cellular redox homeostasis. The Glutathione Peroxidases family, is composed of several paralogs, some of them containing a selenocysteine residue while others are cysteine homologues.
When aligning each protein from the GPx family to Helostoma temminkii’s genome, multiple scaffolds showed significant hits and this is due to the fact that Glutathione Peroxidases are very similar to each other, thus, the reference protein can align to other genomic regions coding for different GPx from the same family. In order to avoid considering that a protein has been duplicated when it actually corresponds to another protein from the same family, a detailed analysis has been carried out in order to select the hits with the highest scores and the lowest E-values for each protein, avoiding the scaffolds that were selected for the others.
GPx1a
GPx1a shows very similar hits when aligning with Gpx1a from Zebrafish (SPP00000621_2.0), however the selected one is OMLM01008245.1, which has the highest score and the lowest E-value.
The gene predicted is located between 30405 and 31613 in the positive strand. Two exons are predicted for this protein with Exonerate, and a Sec is found as expected. Very similar predictions are obtained using both Exonerate and Genewise, although there is one amino acid more in the N-terminal with the Exonerate prediction. The prediction does not include 5 amino acids of the N-terminal nor 3 amino acids of the C-terminal.
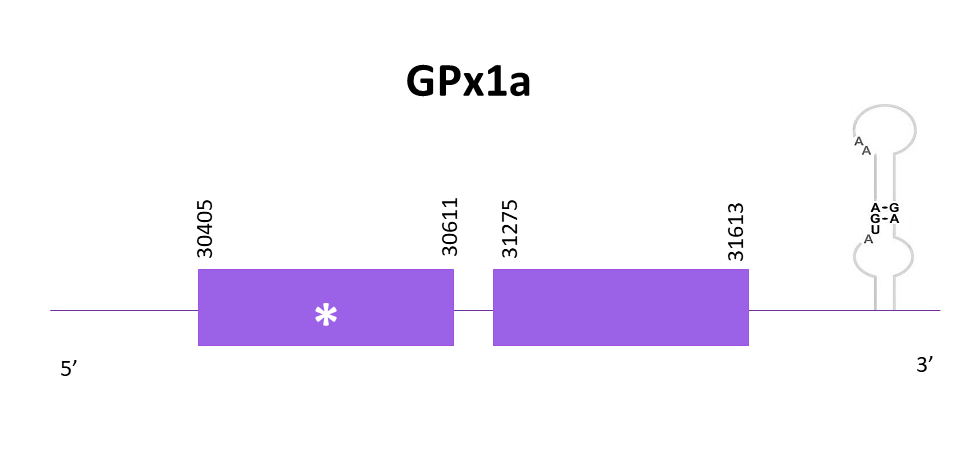
When using Seblastian, the prediction obtained is better taking into account that the N-terminal is complete, even if the C-terminal is still not complete. Also, a SECIS element is predicted in the 3'-UTR region.
GPx1b
Several scaffolds showed significant hits when aligning the corresponding protein from Zebrafish (SPP00000624_2.0) to Helostoma temminkii’s genome. OMLM01004028.1 was the selected scaffold since the one that showed the lowest E-value and highest score was already assigned to GPx1a.
The gene coding for GPx1b is located between 115184 and 116333 positions from the selected scaffold in its negative strand. 2 exons are predicted for this protein using Exonerate and a conserved selenocysteine is detected in the first one. A SECIS structure is also predicted in the 3'-UTR region using Seblastian.
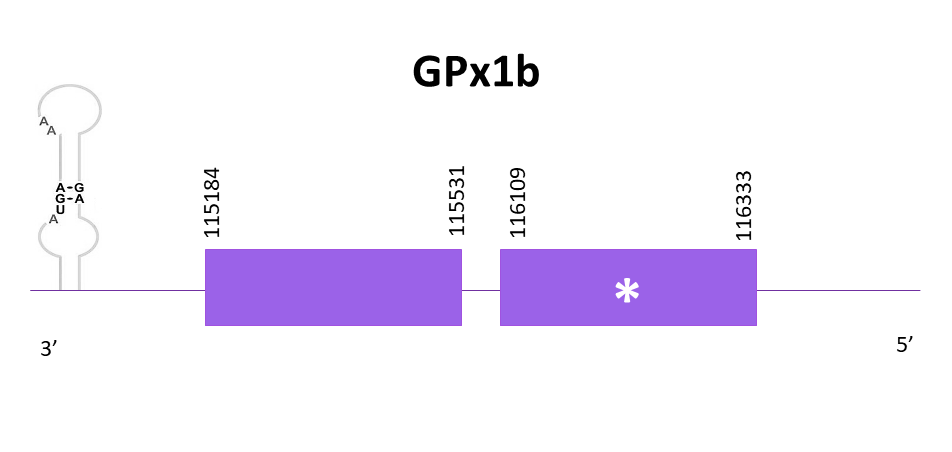
Proteins predicted using Exonerate and Genewise are identical. Both predictions include the initial methionine and preserve the same amino acids in its C-terminal region as in the reference protein. Finally, using Seblastian a prediction of the protein for Helostoma temminkii is also obtained and this one appears to be the same as the other two.
GPx2
OMLM01020689.1 is the scaffold that shows the highest score and lowest E-value as a result of the alignment with the corresponding protein from Zebrafish (SPP00000616_2.0).
The gene for GPx2 is located between 40818 and 41983 positions in the negative strand. 2 exons are predicted for this protein and a selenocysteine can be found in the first one. One SECIS structure is also predicted whereas two SECIS structures are found in GPx2 from Zebrafish and just one in Medaka. Taking into consideration that Helostoma temminkii is more closely related to Medaka rather than to Zebrafish, it makes evolutionary sense to find only one SECIS structure in our species of study.
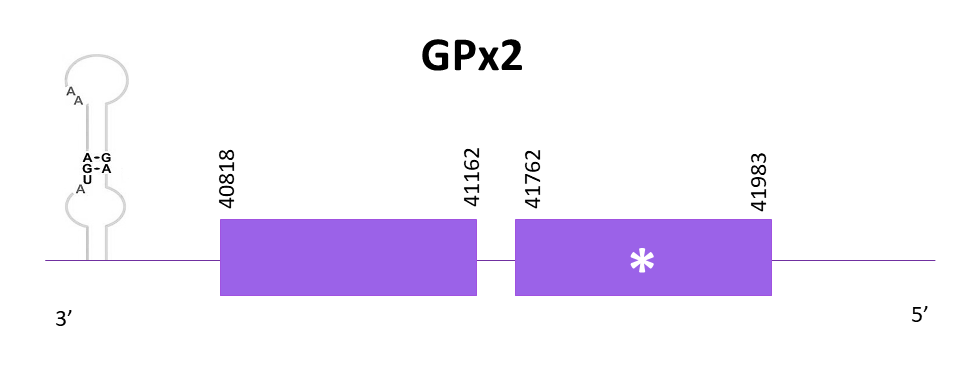
Proteins predicted using Exonerate and Genewise are the same. Both predictions include the initial methionine and preserve the last amino acids in its C-terminal region from the reference protein. Finally, using Seblastian a prediction of the protein for Helostoma temminkii is also obtained and this one appears to be identical to the other two protein predictions.
GPx3a
In this case, the scaffold selected when aligning the corresponding protein from Zebrafish (SPP00000617_2.0) to Helostoma temminkii’s genome is the one with lowest E-value and highest score, OMLM01011369. As expected, there is a selenoprotein.
The gene predicted is located between 2388-3823 in the negative strand. The predicted protein is incomplete compared with the references species, in both C and N-terminal, with both Exonerate and Genewise.
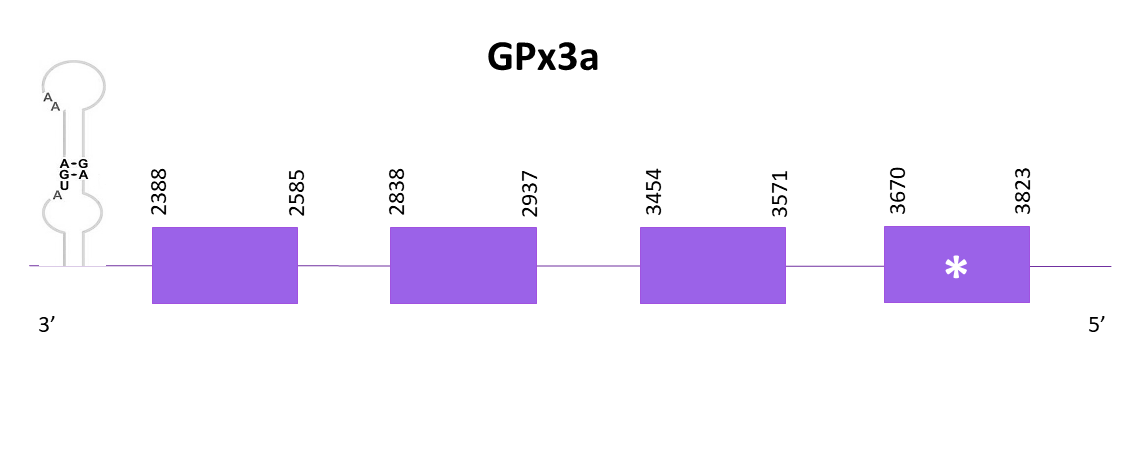
Finally, the prediction obtained with Seblastian is better and more extensive than the previously commented. One predicted SECIS element is found.
GPx3b
Several scaffolds showed significant hits when aligning the corresponding protein from Zebrafish (SPP00000623_2.0) to Helostoma temminkii’s genome. OMLM01006563.1 was the selected scaffold since it is the one that showed lowest E-value and highest score.
The gene coding for GPx1b is located between 77562 and 78936 positions in the negative strand of the selected scaffold. Using Exonerate, 4 exons are predicted for this protein, and a conserved selenocysteine is detected in the first exon.
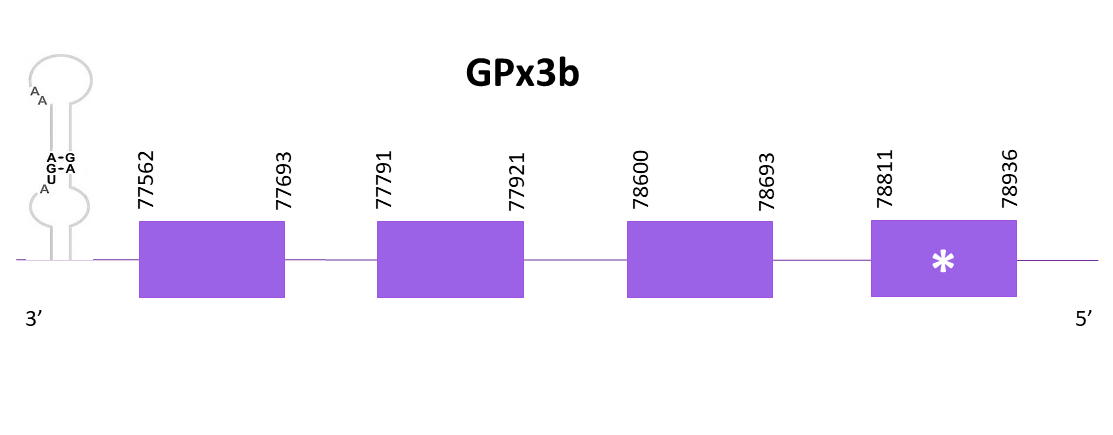
Proteins predicted using Exonerate and Genewise are identical. Both predictions are incomplete in the C-terminal, but complete in the N-terminal.
Finally, using Seblastian there is no selenoprotein detected for this genomic region. Due to that, SECISearch3 is used and one SECIS structure is predicted in the 3'-UTR region.
GPx4a
Different scaffolds showed significant hits when aligning the corresponding protein from Zebrafish (SPP00000618_2.0) to Helostoma temminkii’s genome, and, from these, the one selected (lowest E-value and highest score) was OMLM01007071.1.
The coding gene is located in the positive strand between 36543 and 37350. Using Exonerate, 3 exons are predicted for the protein and a conserved selenocysteine is found in the first exon.
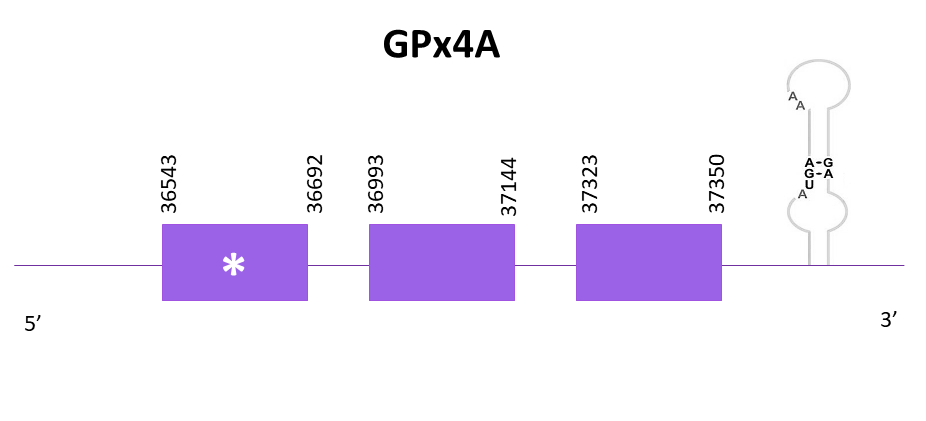
Identical predictions are obtained using Exonerate and Genewise. Both predictions lack some amino acids from the start and preserve the same amino acids in its C-terminal region as in the reference protein.
Finally, using Seblastian a more extensive prediction of the protein for Helostoma temminkii is obtained, including both ends of the protein. A SECIS candidate is predicted in 3'-UTR.
GPx4b
For GPx4b, multiple scaffolds showed significant hits when aligning the corresponding protein from Zebrafish (SPP00000622_2.0) to Helostoma temminkii’s genome, from these, OMLM01021133.1 showed the lowest E-value and highest score and that’s why we selected this one.
The gene goes from position 53146 to 55535 in the negative strand and it consists of 4 exons, the last being the one carrying the selenocysteine. A SECIS structure can be found in the 3'-UTR region of the gene.
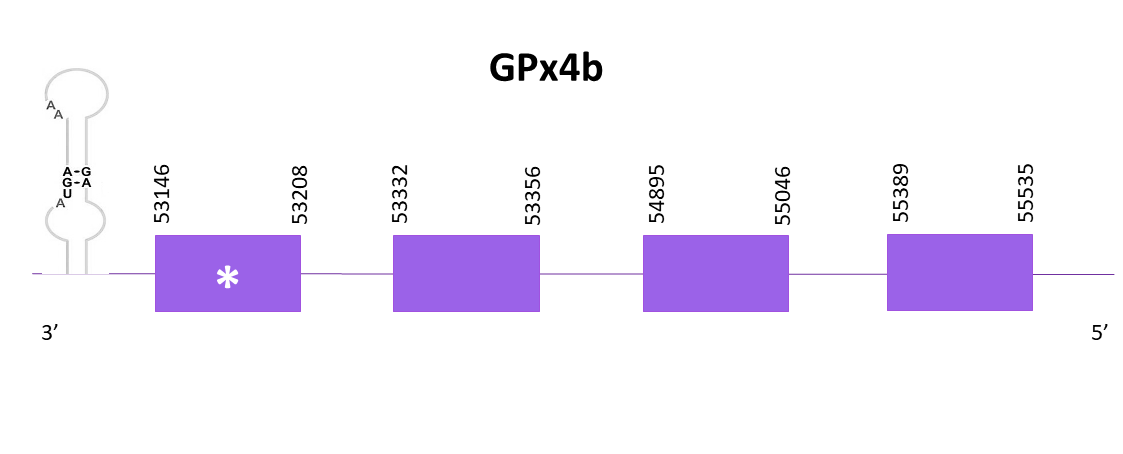
Identical predictions are obtained using Exonerate and Genewise. Both predictions lack the first amino acid from the reference sequence (which is not a methionine and thus we may consider it as incomplete). The predicted protein obtained using Seblastian is more complete than the others on both ends of the protein.
GPx7
Again, following the same criteria, the scaffold selected from the hits from the alignment of Zebrafish (SPP00000619_2.0) and Helostoma temminkii, is OMLM01018267.1. In this case, this protein is a cysteine homolog because it has lost the selenoprotein in both Helostoma temminkii and the reference specie.
The coding gene is located in the negative strand between 14011 and 15778. Using Exonerate, 2 exons are predicted for the protein.
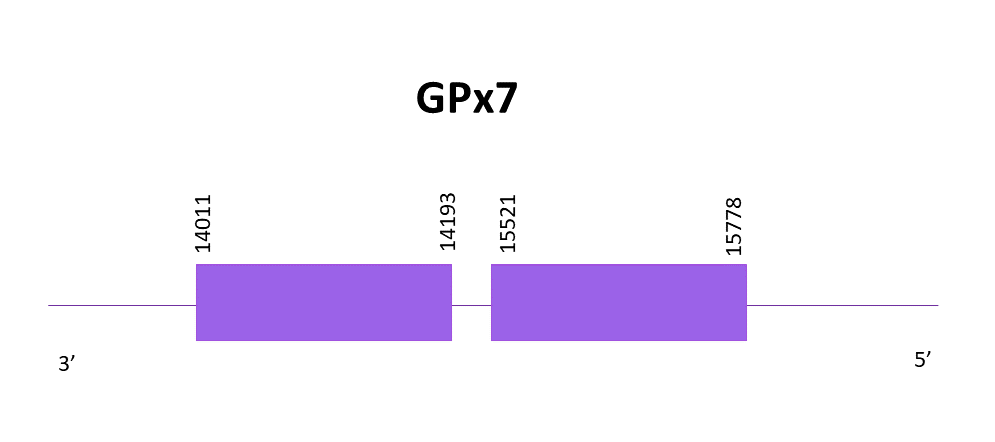
Similar predictions are obtained using Exonerate and Genewise. Both predictions lack a few amino acids from the core of the protein sequence.
Seblastian does not detect any selenoprotein for this genomic region. When using SECISearch3, one SECIS element is predicted but we don’t consider it as valid for our protein due to the fact that it is located in the positive strand and GPx7 is in the negative one.
GPx8
For GPx8, the chosen scaffold is OMLM01014511.1 since it has the highest score and the lowest E-value when aligning the Helostoma temminkii genome to the corresponding protein from Zebrafish (SPP00000620_2.0).
The gene is located in the positive strand, between 4706 and 6520. It does not contain a selenocysteine residue in both Helostoma temminkii and the reference species so it is a cysteine homolog protein, not a selenoprotein. 3 exons are predicted using Exonerate and no SECIS structure is predicted using Seblastian nor SECISearch3.
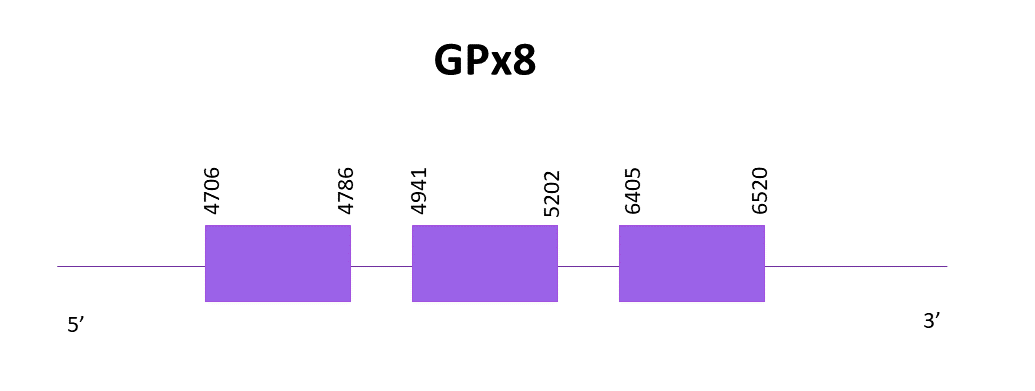
The resulting proteins from Exonerate and Genewise are very similar being an insertion of some amino acids in the Genewise predicted protein the only difference. Both predictions are complete when comparing them to the reference protein from Zebrafish, which does not contain an initial methionine.
After analysing all the selenoproteins found in Helostoma temminkii from the GPx family, we performed a phylogenetic tree to prove that the genomic regions were correctly selected for each protein and to observe which proteins are more closely related within the GPx family.
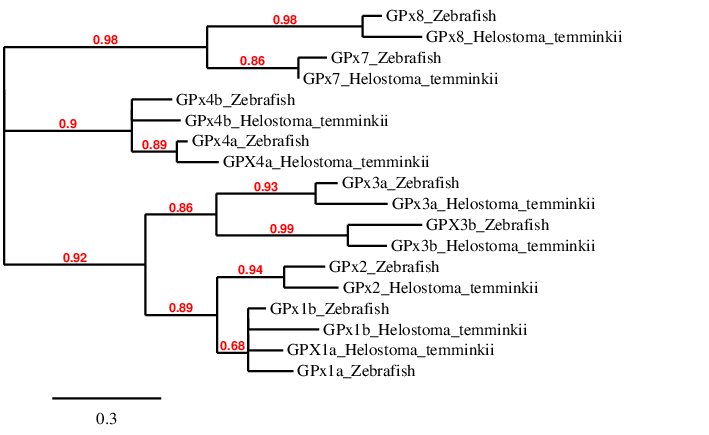
For this phylogenetic tree, we collapsed branches which had a support value smaller than 10%. First of all, we can see that each protein from Helostoma temminkii’s GPx family is grouped together with the corresponding homolog from Zebrafish, proving that we have assigned the correct genomic region from our studied species to each protein. We can observe that GPx7 and GPx8 are more closely related within each other rather than with the rest of paralogs, which makes sense taking into account that these are the only two cysteine homologous proteins while the others are selenoproteins (they contain a selenocysteine residue). It can also be seen that GPx4a and GPx4b are more closely related to each other rather than to the rest of proteins. This can also be seen with GPx1a and GPx1b. Also, GPx2 proteins (from Zebrafish and Helostoma temminkii) have a closer relationship to GPx1 proteins compared to the rest of GPx. Finally, GPx3a proteins from both Zebrafish and Helostoma temminkii are grouped together, the same can be observed for GPx3b proteins; and these two groups of GPx3 proteins are more closely related with each other than to the rest.
As previously explained in the Introduction, Msr A is highly conserved and its main function is to catalyze the enzymatic reduction of the amino acid methionine-S-sulfoxide by using thioredoxin.
MsrA_1
When aligning the corresponding protein from Zebrafish (SPP00000625_2.0) to Helostoma temminkii’s genome, three different scaffolds showed significant hits. OMLM01000544.1 was the best candidate since it had the lowest E-value and highest score, and taking into consideration that another one is the assigned to MsrA_2 while the third one was just a short segment of the protein with a low identity.
The coding gene is located between 1461 and 21097 positions from the scaffold in its positive strand. 5 exons are predicted for this protein using Exonerate and a cysteine is found, like in the reference species.
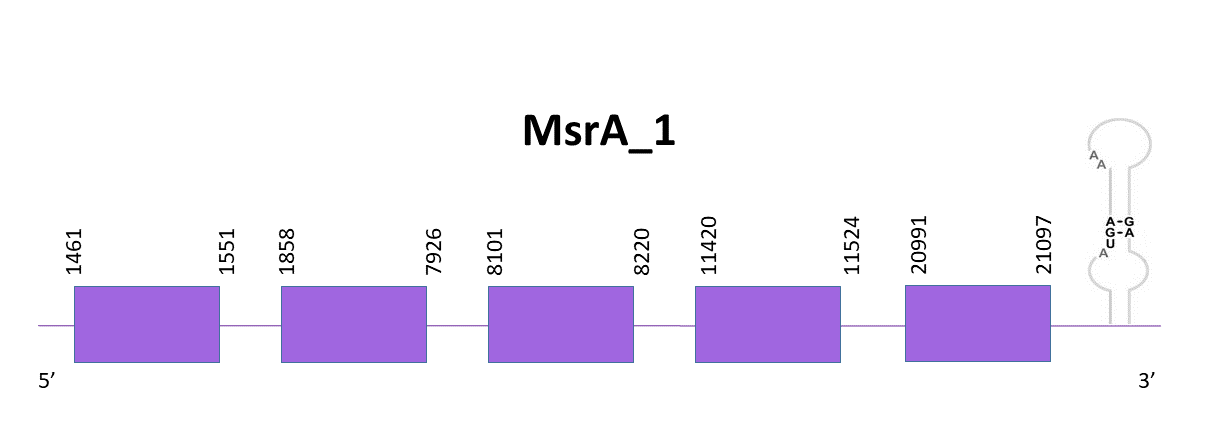
The predictions obtained with Exonerate and Genewise are pretty similar, although the one obtained with Exonerate presents a few more amino acids. Both predictions include the initial methionine. Finally, using Seblastian there is no selenoprotein nor SECIS detected for this genomic region. When using SECISearch3, one SECIS element is predicted, approximately 200 nucleotides away from the coding gene on its 3'-UTR end. We believe that this would be a false positive SECIS prediction since the SECIS structure would be very distant to the rest of the gene and both reference species (Medaka and Zebrafish) also present no SECIS.
MsrA_2
Four different scaffolds showed significant hits when aligning the corresponding protein from Zebrafish (SPP00000626_2.0) to Helostoma temminkii’s genome, but following the same criteria as before, the one selected was OMLM01009250.1.
The gene coding for the MsrA_2 protein is located between 119818 and 134704 positions from the scaffold in its negative strand. 5 exons are predicted for this protein using Exonerate and a cysteine is found, like in the reference species.
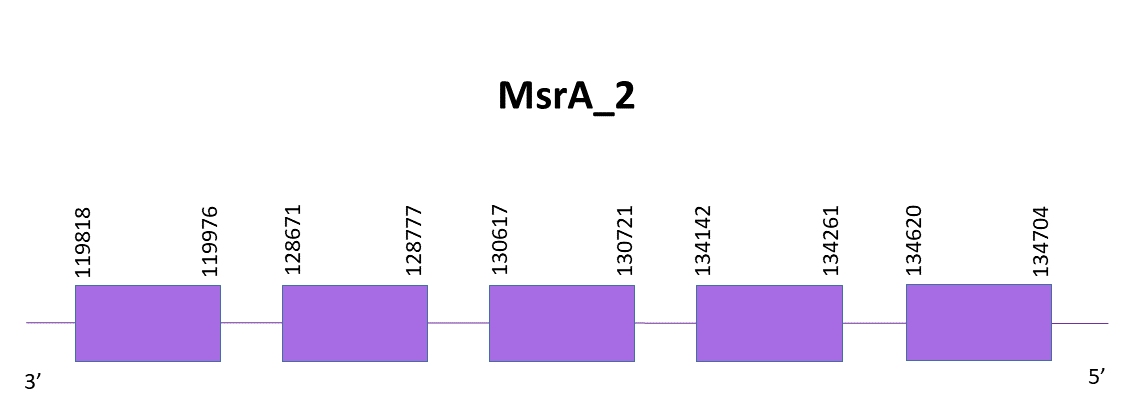
Identical predictions are obtained with Exonerate and Genewise. Both predictions lack some amino acids like the initial Met.
Finally, using Seblastian there is no selenoprotein detected for this genomic region, and when using SECISearch3, no SECIS element is predicted as expected.
After analysing the MsrA proteins found in Helostoma temminkii’s genome, we performed a phylogenetic tree to prove that the genomic regions were correctly selected for each protein and to establish evolutionary relationships among species.
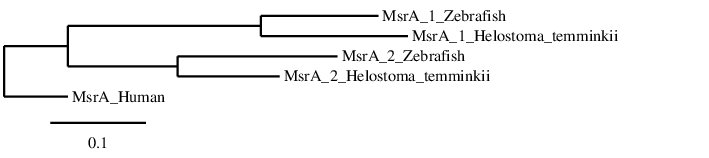
We rerooted this phylogenetic tree in order to select the human MsrA as the outgroup, since we know that Homo sapiens is the most evolutionary distant species from the studied ones. First of all, we can see that each protein from Helostoma temminkii’s MsrA family is grouped together with the corresponding homolog from Zebrafish, proving that we have assigned the correct genomic region from our studied species to each protein. Finally, it can be seen that the human MsrA is separated from the fishes’ MsrA and the duplication is only present in fishes, probably as a result of the whole-genome duplication event from bony fishes (Ts3R).
Methionine-R-sulfoxide reductases (MSRB) are zinc-containing selenoproteins that were previously identified as Selenoproteins R (SelR) and Selenoprotein X (SelX). Several homologs (MSRB1a, MSRB1b, MSRB2 and MSRB3) have been annotated in Danio rerio.
MSRB1a
Two different scaffolds showed significant hits when aligning the corresponding protein from Zebrafish (SPP00000645_2.0) to Helostoma temminkii’s genome. OMLM01007873.1 was the best candidate since it had the lowest E-value and highest score and the other scaffold corresponds to MSRB1b, since both proteins are very similar they align to both genomic regions.
The gene coding for MSRB1a is located between 24155 and 25304 positions from the scaffold in its positive strand. 3 exons are predicted for this protein using Exonerate and a conserved selenocysteine is detected in the last exon. Two SECIS structures are predicted in the 3'-UTR region.
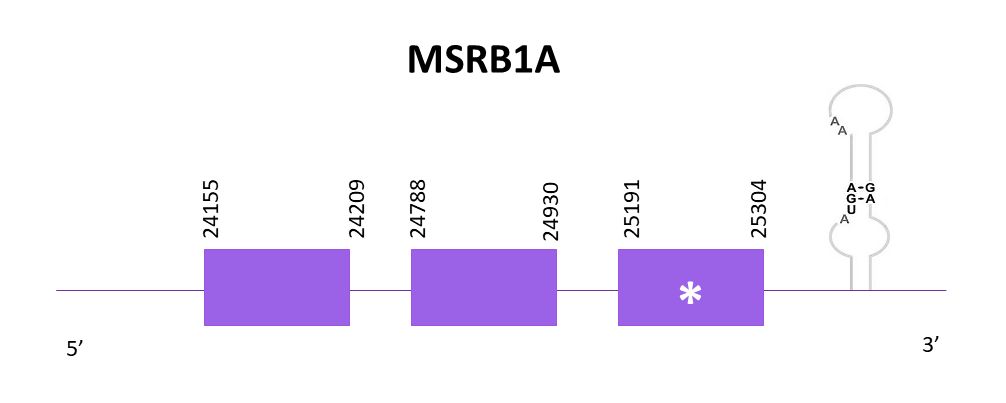
Proteins predicted using Exonerate and Genewise are pretty similar, both of them including the initial methionine and showing some gaps in their T-coffee alignments with the reference protein at the C-terminal region. Finally, using Seblastian a prediction of the protein for Helostoma temminkii is also obtained and this one seems to be better than the others because it is more complete (it fills the gaps that were present in the previous ones).
MSRB1b
For MSRB1b, two scaffolds showed significant hits when aligning the corresponding protein from Zebrafish (SPP00000644_2.0) to Helostoma temminkii’s genome, from which the selected one was OMLM01020828.1, due to the fact that the other scaffold is the one analysed in MSRB2.
The gene goes from position 15774 to 16886 in the positive strand and with Exonerate 3 exons are found. There is a conserved selenocysteine in the third exon. A SECIS structure can be found in the 3'-UTR region of the gene.
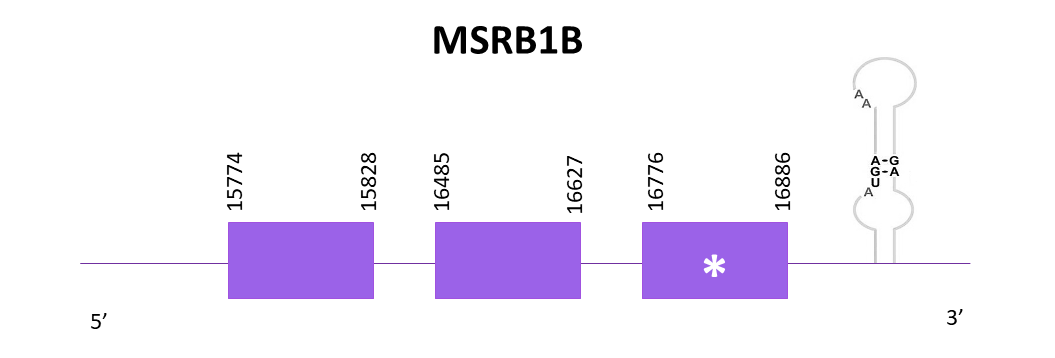
The predictions obtained using Exonerate and Genewise are identical. Both predictions include the first amino acid but lack the last one. The predicted protein obtained using Seblastian is more complete than the others on both ends of the protein.
MSRB2
For MSRB2, the chosen scaffold is OMLM01005029.1 since it has the highest score and the lowest E-value when aligning the Helostoma temminkii genome to the corresponding protein from Zebrafish (SPP00000646_2.0).
The gene is located in the positive strand, between 50653 and 53160. There is a cysteine homolog protein, in this species as well as in the reference species. 5 exons are predicted using Exonerate and no SECIS structure is predicted using Seblastian nor SECISearch3.
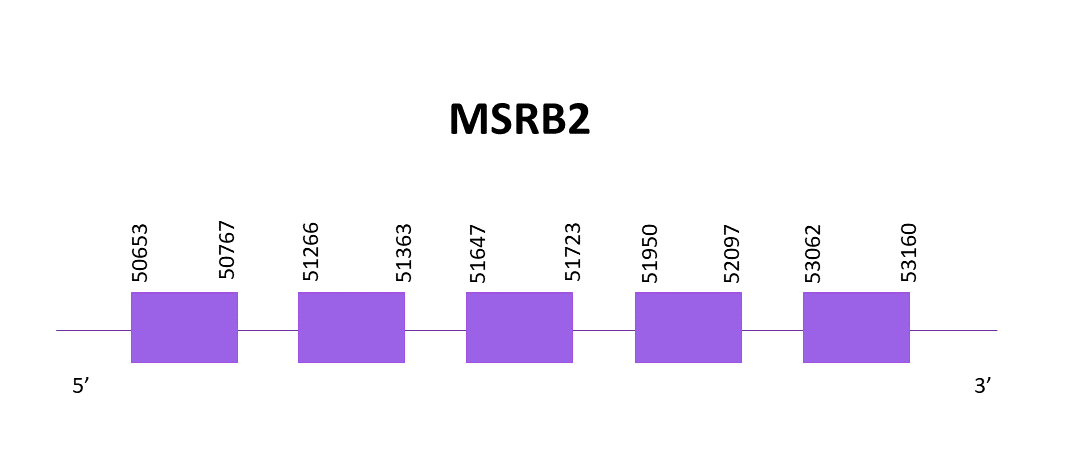
The resulting predictions from Exonerate and Genewise are identical. Both contain an initial methionine and lack the last amino acid.
MSRB3
In this case, three scaffolds showed significant hits when aligning the corresponding protein from Zebrafish (SPP00000647_2.0) to Helostoma temminkii’s genome, from these, OMLM01009714.1 showed the lowest E-value and the highest score and that’s why we selected this one, the other two scaffolds presented shorter hits aligned and with lower identity values.
The gene goes from position 953 to 2615 in the positive strand and it consists of 5 exons, the protein does not contain a selenocysteine, thus being a cysteine homologue. No SECIS structure can be found, which concur with MSRB3 from Zebrafish.
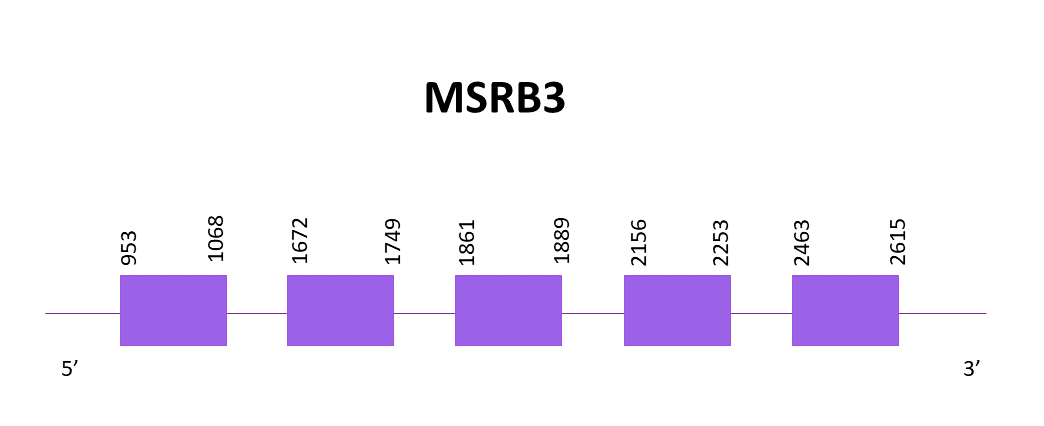
Both predictions obtained using Exonerate and Genewise lack part of the initial and final part of the protein, being the central region of the protein the one that is better aligned.
After analysing the proteins found in Helostoma temminkii from the MSRB family, we performed a phylogenetic tree to prove that the genomic regions were correctly selected for each protein and to establish evolutionary relationships among species.
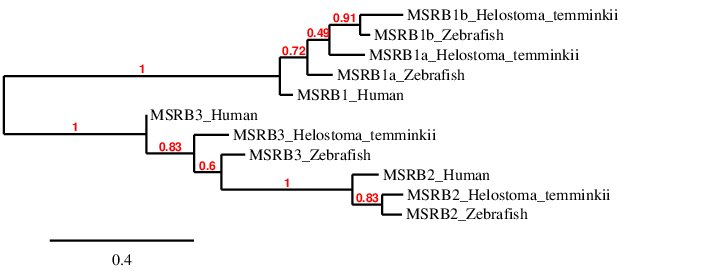
First of all, we can see that each protein from Helostoma temminkii’s MSRB family is grouped together with the corresponding homolog from Zebrafish, proving that we have assigned the correct genomic region from our studied species to each protein. Also, it can be seen that proteins predicted for Helostoma temminkii are more closely related to Zebrafish rather than to Human, which makes evolutionary sense. We can also point out that MSRB2 proteins are more similar to MSRB3 rather than to MSRB1, probably indicating that this protein was generated from a duplication taking place later in time. This is also consistent with the fact that both MSRB2 and MSRB3 are cysteine homologues whereas MSRB1s are selenoproteins. Finally, it can be seen that the human MSRB1 is separated from the fishes’ MSRB1 which are grouped together (MSRB1a and MSRB1b) and the duplication is only present in fishes, probably as a result of the whole-genome duplication event from bony fishes (Ts3R).
Sel15, which is also known as Sep15 in mammals, together with Selenoprotein M is a thioredoxin-like fold ER-resident protein.
Again, following the same criteria, the scaffold selected from the hits from the alignment of Zebrafish (SPP00000632_2.0) and Helostoma temminkii, is OMLM01002590.1. In this case, this protein contains a selenocysteine in both Helostoma temminkii and the reference species.
The coding gene is located in the positive strand between 127859 and 132270. Using Exonerate, 4 exons are predicted for the protein.
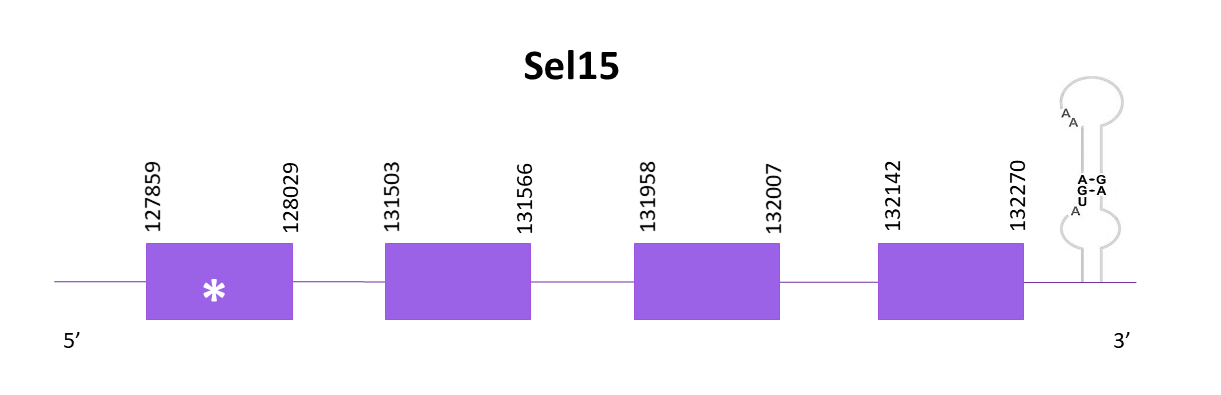
Identical predictions are obtained using Exonerate and Genewise, both lacking 8 amino acids from the start of the protein.
The prediction obtained with Seblastian has the complete protein. One SECIS element is predicted using Seblastian.
SELENOE has only been detected in fish, its location is inside the ER and its function remains unknown.
OMLM01000156.1 is the chosen scaffold since it is the only one with significant hits when aligned to the corresponding SELENOE from Zebrafish (SPP00000615_2.0).
The gene coding for SELENOE is located between 88242 and 90050 positions from the selected scaffold in its positive strand. 4 exons are predicted for this protein using Exonerate and a conserved selenocysteine is detected in the second exon. A SECIS structure is predicted in the 3'-UTR region.
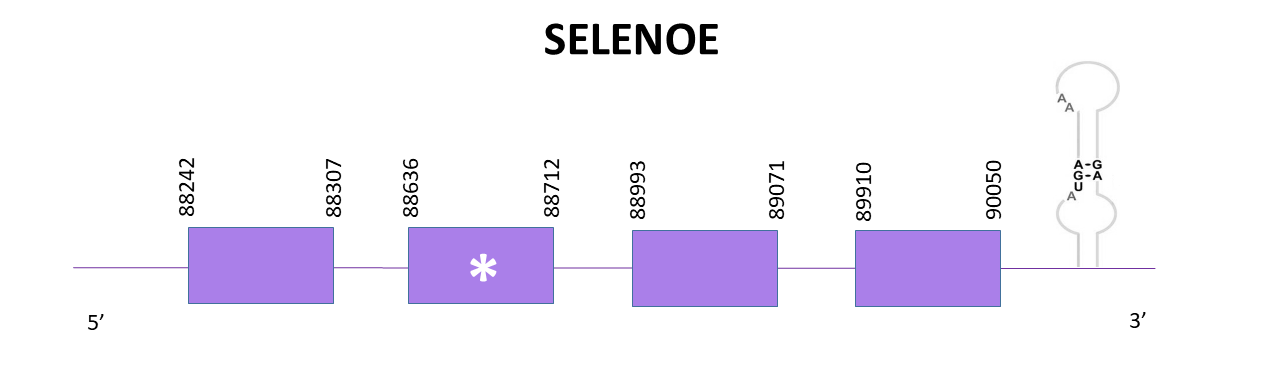
Proteins predicted using Exonerate and Genewise lack the N-terminal end (being the prediction from Exonerate a bit more extended if compared to the one from Genewise) whereas we obtain the full protein (starting with methionine) using Seblastian.
SELENOH is a stress-related selenoprotein that acts as an oxidoreductase.
When aligning the reference species (SPP00000634_2.0) with Helostoma temminkii only one significant hit is obtained, so the scaffold selected is OMLM01018966.1.
The gene goes from 28565 to 30446 in the negative strand. According to Exonerate, there are 3 exons and a selenocysteine is conserved. A SECIS element is predicted in 3'-UTR.
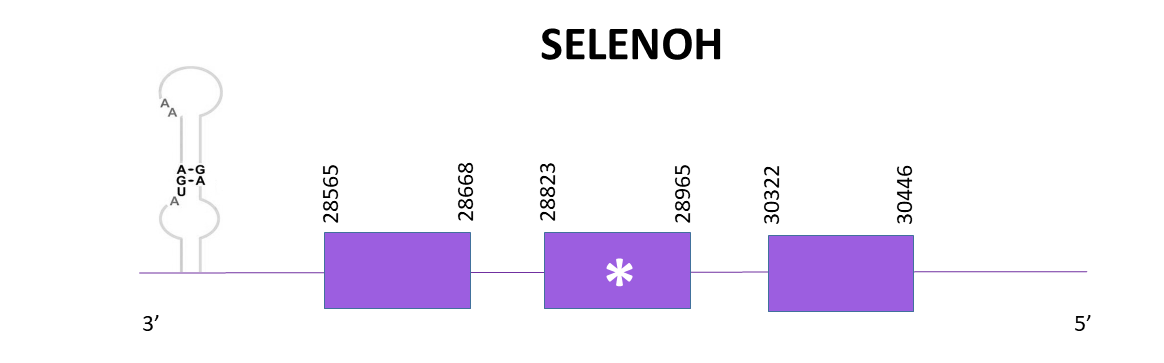
The prediction with Exonerate is more complete than the one with Genewise, it only lacks the 3 first amino acids while the one obtained with Exonerate lacks more amino acids. The prediction using Seblastian differs in the start with the one obtained with Exonerate, taking into consideration that the Exonerate one starts with Met and the Seblastian one does not.
SELENOI is a transmembrane protein which can only be found in vertebrates.
For SELENOI, the chosen scaffold is OMLM01006824.1 since it has the highest score and the lowest E-value when aligning the Helostoma temminkii genome to the corresponding protein from Zebrafish (SPP00000635_2.0). The other two scaffolds with significant hits were poorly aligned and only representing a small fragment of the protein.
The gene predicted is located between 68289 and 75601 in the negative strand. Ten exons are predicted for this protein with Exonerate, and a selenocysteine is found in the last exon. Also, a SECIS is predicted at the 3'-UTR region using Seblastian.
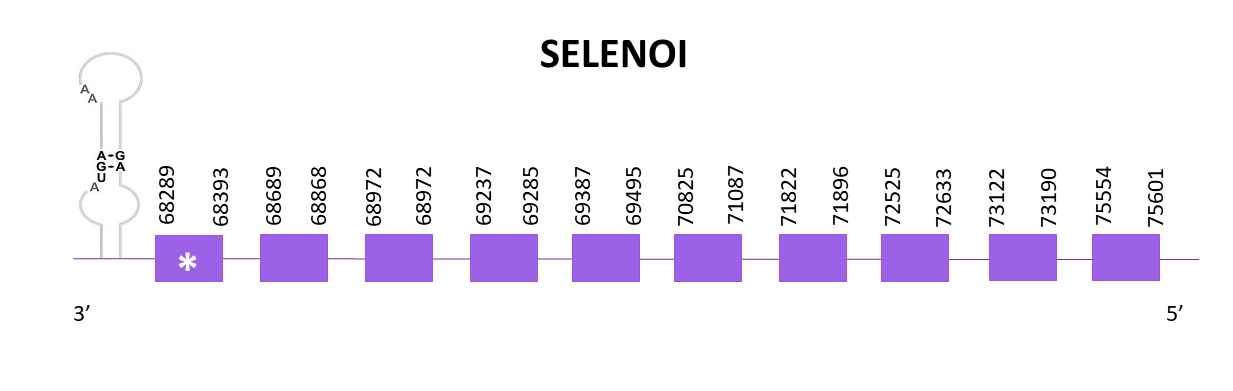
The predicted protein obtained using Exonerate shows a very good alignment in T-Coffee with the reference protein whereas the one from Genewise is incomplete at the C-terminal end (missing 14 amino acids). Both of them do not have an initial methionine and the reference protein neither.
When using Seblastian, the prediction obtained is better taking into account that the protein begins with a methionine but the last fragment of the protein is still not predicted.
SELENOJ is only found in actinopterygian fishes and sea urchin, but not in other vertebrates (mammals, birds and amphibians). The potential role of of Selenoprotein J is during embryogenesis, preferentially expressed in the eye lens (it shares significant similarity with jellyfish crystallins).
SELENOJ1
Two scaffolds showed significant hits when aligning the corresponding protein from Zebrafish (SPP00000636_2.0) to Helostoma temminkii’s genome. Both hits show high scores and low E-values, so we analyse both, the first one, OMLM01009182.1, as SELENOJ1, and the second one as SELENOJ1.2 (from highest to lowest score and from lowest to highest E-value).
The gene goes from position 92932 to 95436 in the negative strand and it is composed of 9 exons, one of them carrying the selenocysteine. A SECIS structure can be found in the 3'-UTR region of the gene, nevertheless, in the reference species no SECIS can be found.
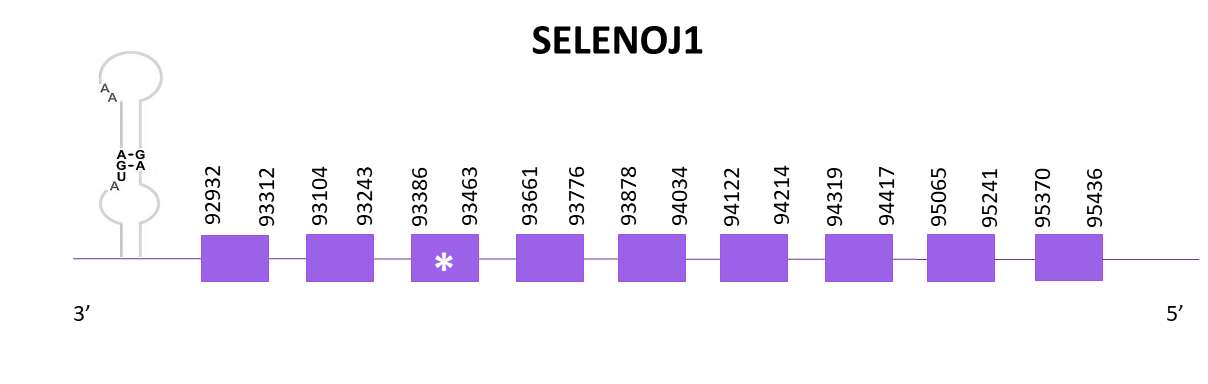
Identical predictions are obtained using Exonerate and Genewise. Both predictions include the Met and lack 6 amino acids of the C end. The predicted protein obtained using Seblastian is equally complete as the previously obtained, but includes the 6 amino acids.
SELENOJ1.2
As previously commented, the scaffold selected for SELENOJ1.2 is OMLM01014617.1.
The coding gene from position 36606 to 42315 in the negative strand. According to the Exonerate there are 8 exons, one of them carrying the selenocysteine. A SECIS structure can be found in the 3'-UTR region of the gene, nevertheless, in the Zebrafish no SECIS can be found, so we compared our species with Medaka, which has a SECIS element. Taking into consideration that Helostoma temminkii is more closely related to Medaka rather than to Zebrafish, it makes evolutionary sense to find only one SECIS structure in our species of study.
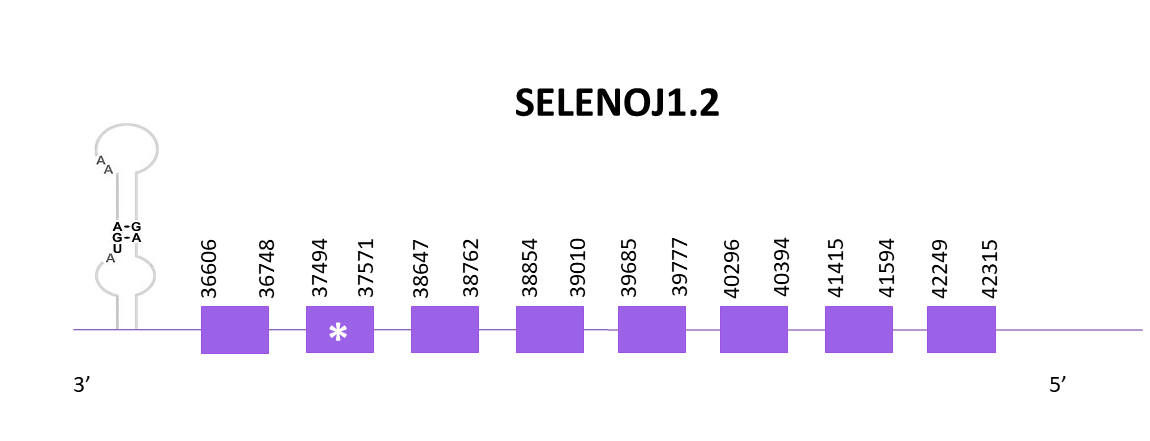
Identical predictions are obtained using Exonerate and Genewise, except for the last part of the protein where Exonerate lacks the last amino acids. The predicted protein obtained using Seblastian is equally complete as the previously obtained, but includes the 6 amino acids that were lacking before.
After analysing the selenoproteins found in Helostoma temminkii corresponding to SELENOJ we suspect that a duplication took place, we performed a phylogenetic tree to prove that our hypothesis was correct.
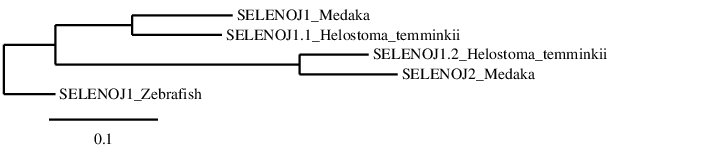
For this phylogenetic tree, we included the SELENOJ1 from Zebrafish as well as SELENOJ1 and SELENOJ2 present in Medaka. The obtained phylogenetic tree proves that Helostoma temminkii has a duplication of SELENOJ on its genome, being the protein that we named as SELENOJ1.1 homologous to SELENOJ1 from Medaka and SELENOJ1.2 homologous to SELENOJ2. It can also be observed that SELENOJ1 from Zebrafish is more distant to the other proteins, probably indicating that the duplication event occurred subsequent to the divergence of Zebrafish from the other lineages.
SELENOK is known to be related with SelS based on their topology.It is involved in mechanisms in charge of endoplasmic reticulum associated degradation of proteins that are glycosylated and misfolded.
The scaffold selected is the only hit from the alignment of Zebrafish (SPP00000633_2.0) and Helostoma temminkii, OMLM01000630.1. This protein contains a selenocysteine in both Helostoma temminkii and the reference species.
The coding gene is located in the positive strand between 1212 and 2975. Using Exonerate, 4 exons are predicted for the protein.
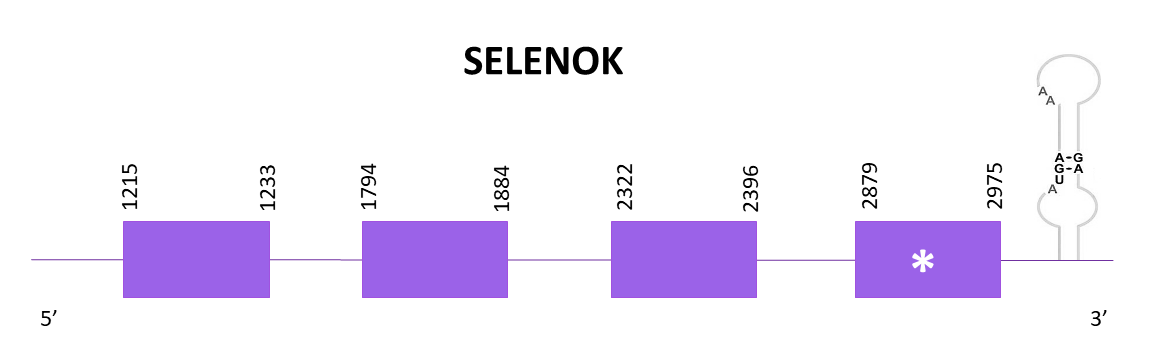
Similar predictions were obtained with Exonerate and Genewise, nevertheless, Genewise does not align the last 3 amino acids and Exonerate does. The prediction obtained with Seblastian has the complete protein, including the first Met. One SECIS element is predicted using Seblastian, as expected according to the presence of SECIS in both Medaka and Zebrafish.
SELENOL contains two selenocysteine residues and it is restricted to aquatic organisms.
No significant hits were obtained for any scaffold when aligning the Helostoma temminkii’s genome with the corresponding protein from Zebrafish (SPP00000637_2.0). The E-values of the scaffolds obtained were higher than the threshold that we established for all the proteins. For this reason, we believe that SELENOL is not present in Helostoma temminkii’s genome.
SELENOM is (together with Sel15) a thioredoxin-like folds ER-resident protein and its function remains unknown.
Two scaffolds showed significant hits when aligning the corresponding protein from Zebrafish (SPP00000638_2.0) to Helostoma temminkii’s genome. We selected the scaffold which had the highest score and the lowest E-value (OMLM01020340.1) since the other one had low score and identity values and was only aligned to a small portion of the protein.
The gene predicted is located between 70537 and 73037 in the positive strand. Five exons are predicted for this protein using Exonerate, and a selenocysteine is found. Also, a SECIS is predicted at the 3'-UTR region using Seblastian.
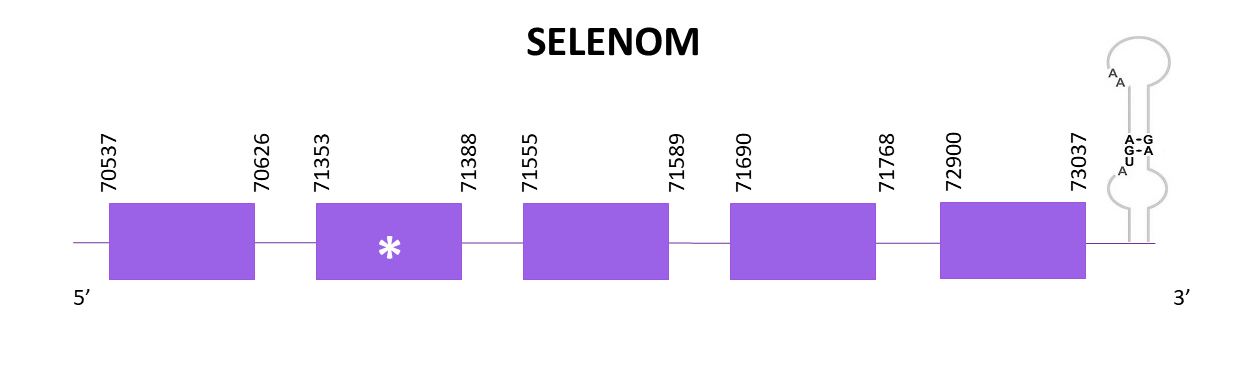
The predicted protein obtained using Exonerate shows a very good alignment in T-Coffee with the reference protein whereas the one from Genewise is incomplete at the N-terminal end (missing aproximately 40 amino acids). Both of them do not have an initial methionine.
When using Seblastian, the prediction obtained is better taking into account that the protein begins with a methionine and the last fragment of the protein is better predicted.
SELENON is a glycoprotein localized in the endoplasmic reticulum. It is enrolled in redox regulation of calcium homeostasis and in cell protection against oxidative stress.
When aligning the corresponding protein from Zebrafish (SPP00000639_2.0) to Helostoma temminkii’s genome, only one scaffold showed a significant hit, OMLM01014045.1.
The coding gene is located between 24931 and 29638 positions from the scaffold in its negative strand. 12 exons are predicted for this protein using Exonerate and a selenocysteine is found, like in the reference species.
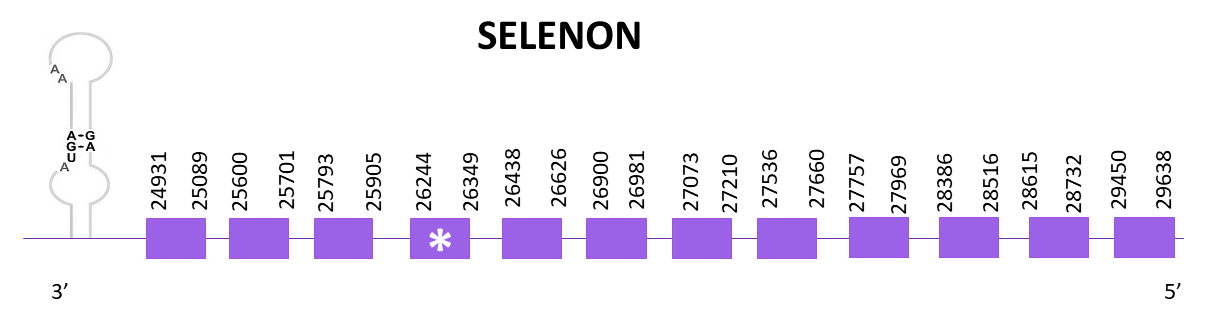
The predictions obtained with Exonerate and Genewise are identical. Both predictions lack the last amino acid but include the initial Met. Finally, using Seblastian the prediciton obtained is slightly better taking into account that it includes the two amino acids lacking in the previous predicitons. One SECIS element is predicted in the 3'-UTR end, as expected.
SELENOO was discovered more than a decade ago but no structural or biochemical characterization of this protein has been reported. It is the largest selenoprotein present in mammals.
SELENOO1
OMLM01015002.1 is the chosen scaffold, following the same criteria as previously mentioned, from the significant hits obtained when aligned to the corresponding protein from Zebrafish (SPP00000640_2.0) with Helostoma temminkii.
The gene coding is located between 32365 and 38561 positions from the selected scaffold in its negative strand. 9 exons are predicted for this protein using Exonerate and a conserved selenocysteine is detected. A SECIS structure is predicted in the 3'-UTR region as expected (like the Zebrafish).
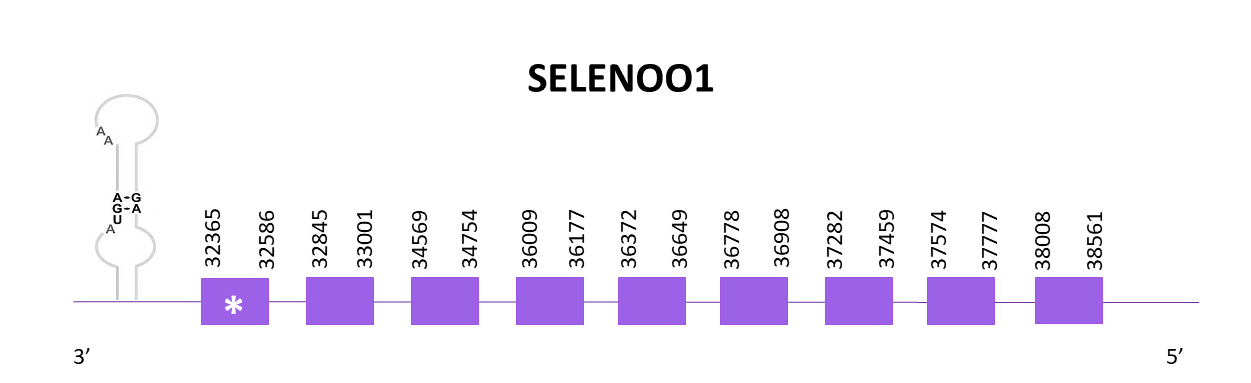
The predictions obtained with Genewise and Exonerate are almost identical, the only difference is that the Genewise one lacks the last 3 amino acids. The Exonerate prediction is complete, starting with the Met, and it is the same prediction as the one obtained using Seblastian.
SELENOO2
Three different scaffolds showed significant hits when aligning the corresponding protein from Zebrafish (SPP00000641_2.0) to Helostoma temminkii’s genome, and the one selected was OMLM01014659.1. The first scaffold had a low identity, the second one is the best match for SELENOO1, so the third one is the one selected.
The gene coding for the SELENOO2 protein is located between 12499 and 16682 positions from the scaffold in its positive strand. 10 exons are predicted for this protein using Exonerate and a selenocysteine is found, like in the reference species.
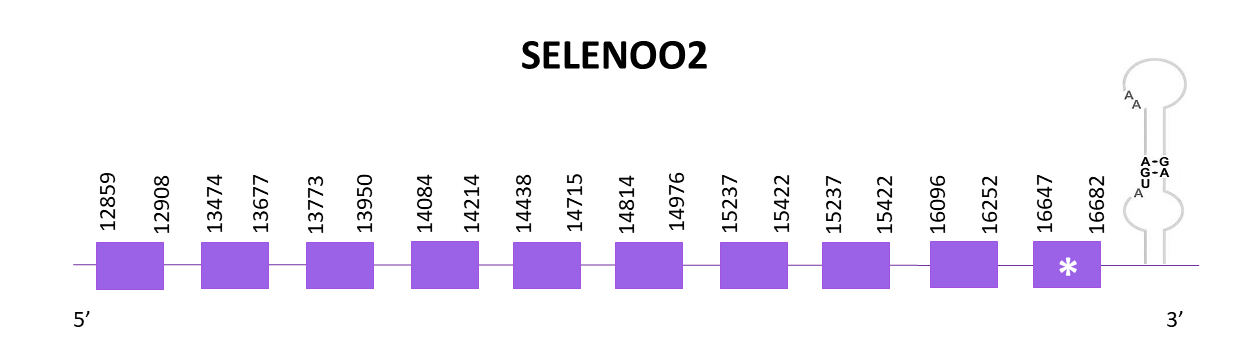
The protein prediction obtained with both Exonerate lacked the first 14 amino acid and the 64 of the end, while the Genewise prediction lacked the 11 amino acid of the start, and the same 64 of the end. Both did not include Met.
Finally, using Seblastian there is no selenoprotein detected for this genomic region, and when using SECISearch3, one SECIS element is predicted as expected.
After analysing the selenoproteins found in Helostoma temminkii for SELENOO, we performed a phylogenetic tree to observe the evolutionary relationships among species.

For this phylogenetic tree we can see that Homo sapiens is the most evolutionary distant species from the studied ones. It can also be seen that SELENOO1 from Helostoma temminkii is grouped together with the corresponding homolog from Zebrafish and the same happens for SELENOO2, proving that we have assigned the correct genomic region from our studied species to each paralog protein. Also, it can be seen that proteins predicted for Helostoma temminkii are more closely related to Zebrafish rather than to Human, which makes evolutionary sense since Human lineage diverged prior in time from the other lineages. This idea is also consistent with the duplication of SELENOO only being present in fishes, probably as a result of the whole-genome duplication event from bony fishes (Ts3R).
This protein has recently evolved and, for that reason, SelP homologous are found predominantly in vertebrates. It is implicated as an extracellular antioxidant and also in the selenium transport to extra-hepatic tissues.
SELENOP_1
When aligning the protein of Zebrafish (SPP00000642_2.0) and Helostoma temminkii, two scaffolds had significant hits. The one selected has the higher score and lowest E-value, OMLM01003153.1, while the other one is the one assigned to SELENOP_2. This protein contains a selenocysteine in Helostoma temminkii as well as in the two reference species (Medaka and Zebrafish). One SECIS element is predicted in 3'-UTR, as expected taking into account that Medaka also had a SECIS element. Nevertheless, Zebrafish showed no SECIS, that is the reason of the comparison with the second reference species.
The coding gene is located in the negative strand between 17918 and 20472. Using Exonerate, 4 exons are predicted for the protein.
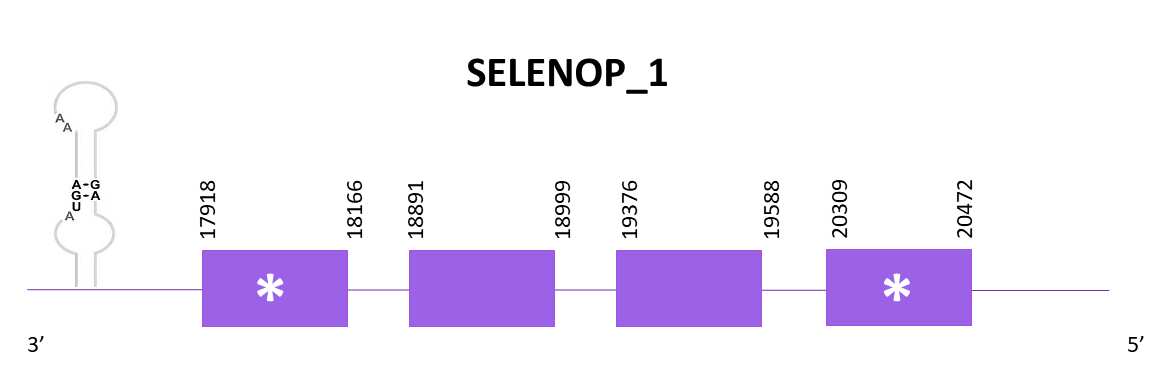
The predictions obtained with Exonerate and Genewise had gaps in both ends of the protein sequence, nevertheless the loss in Exonerate is bigger. The prediction obtained with Seblastian has the complete protein, including the first Met.
SELENOP_2
Two scaffolds showed significant hits when aligning the corresponding protein from Zebrafish (SPP00000643_2.0) to Helostoma temminkii’s genome. We selected the scaffold which had the highest score and the lowest E-value (OMLM01003444.1).
The gene predicted is located between 7532 and 8721 in the negative strand. 4 exons are predicted for this protein with Exonerate, and 1 selenocysteine is found. Also, a SECIS is predicted at the 3'-UTR region using Seblastian.
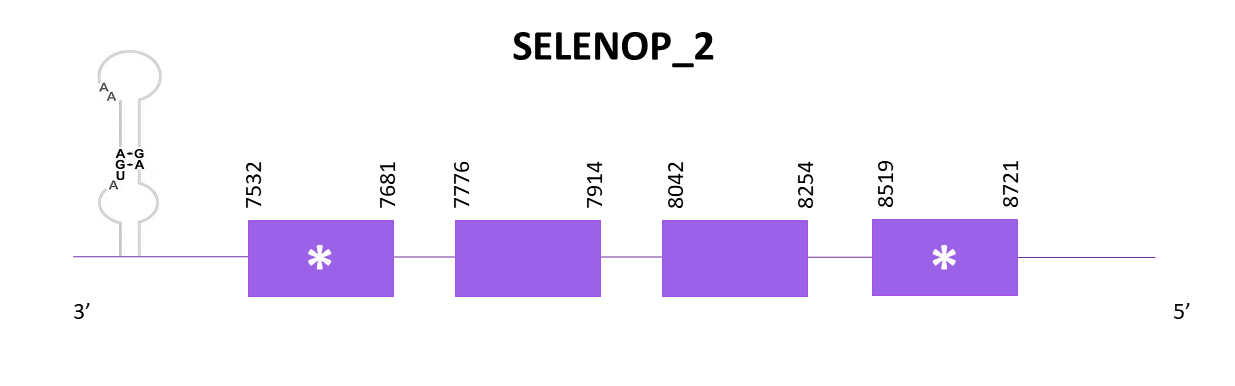
The predicted protein obtained using Exonerate shows a good alignment of the first part of the protein (it starts with Met) but the rest of the protein presents many gaps. When using Genewise, it happens the same as with Exonerate but with even more gaps. Even when using Seblastian, the prediction obtained is not complete.
This selenoprotein is unique because it contains 17 Sec residues per protein in zebrafish, nevertheless, due to the fact that the predicted protein is far shorter than the reference protein, only one selenocysteine is found. However, since Medaka is closely related to Helostoma temminkii and it has 16 selenocysteine, we performed the same search for the SELENOP_2 in Helostoma temminkii but using Medaka’s protein as a reference instead in order to find a better prediction.
SELENOP_2_Medaka
As previously commented, the scaffold selected is OMLM01003444.1.
The gene predicted is located between 6998 and 8721 in the negative strand. 5 exons are predicted for this protein with Exonerate, and 14 selenocysteines are found. In this case, there is also a SECIS candidate predicted at the 3'-UTR region using Seblastian.
The predicted protein with Exonerate and Genewise has several gaps but it is better than the prediction obtained when comparing with Zebrafish. When using Seblastian, the prediction obtained is even better taking into account that it is larger and it includes the 10 selenocysteines. Taking this into account, we conclude that the evolutionary closer relationship with Medaka allows a better alignment and prediction of SELENOP_2 protein.
After analysing the selenoproteins found in Helostoma temminkii for SELENOP, we performed a phylogenetic tree to prove that the genomic regions were correctly selected for each protein and to observe evolutionary relationships among species.
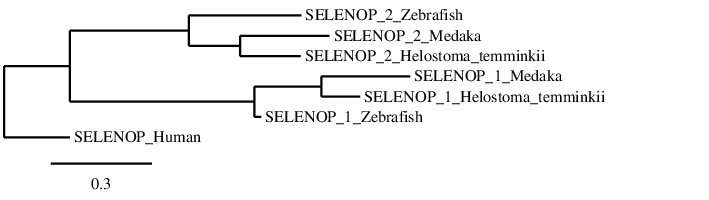
We rerooted this phylogenetic tree in order to select the human SELENOP as the outgroup, since we know that Homo sapiens is the most evolutionary distant species from the studied ones. First of all, we can see that each SELENOP from Helostoma temminkii is grouped together with the corresponding homologues from Medaka and Zebrafish, proving that we have assigned the correct genomic region from our studied species to each paralog protein. Also, it can be seen that proteins predicted for Helostoma temminkii are more closely related to Medaka rather than to Zebrafish, which makes evolutionary sense since Zebrafish diverged prior in time from the other lineages. Finally, it can be seen that the human SELENOP is separated from the fishes’ SELENOPs which are grouped together and the duplication of this protein is only present in fishes, probably as a result of the whole-genome duplication event from bony fishes (Ts3R).
SELENOS encodes for a transmembrane protein that is localized in the endoplasmic reticulum, it is involved in the degradation process of misfolded proteins.
Just one scaffold showed significant hits when aligning the corresponding protein from Zebrafish (SPP00000648_2.0) to Helostoma temminkii’s genome, OMLM01015868.1.
The gene coding for SELENOS is located between 39217 and 43191 positions from the selected scaffold in its negative strand. 8 exons are predicted for this protein using Exonerate and a conserved selenocysteine is detected in the first exon. 3 SECIS structures are also predicted using SECISearch3.
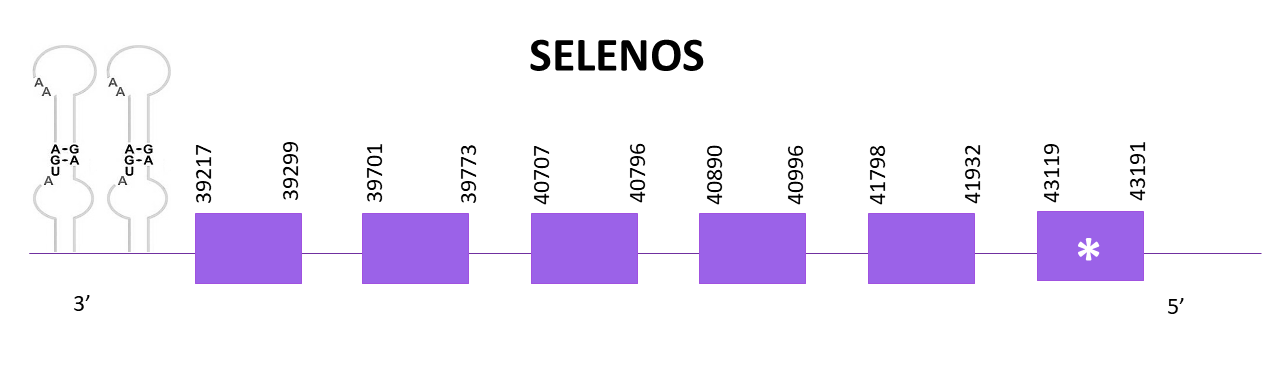
Proteins predicted using Exonerate and Genewise are almost identical, only differing on the last amino acids of the protein. Both predictions lack the first 3 amino acids including the initial methionine. Despite that, we can say that the prediction is almost complete although it includes some amino acid changes.
SELENOT or SelT belongs to the Rdx family of selenoproteins, and, as the name of the family already indicates, it possesses a thioredoxin-like fold and a conserved CxxU motif.
SELENOT1
Two scaffolds had significant hits when comparing with Zebrafish (SPP00000649_2.0). One is discarded because of the low identity, and the other one is selected, OMLM01001838.1 due to the high score and low E-value.
The coding gene for SELENOT1 is located in the negative strand between 89446 and 92565. When using Exonerate 5 exons are predicted and a selenocysteine is found (like in the reference species).
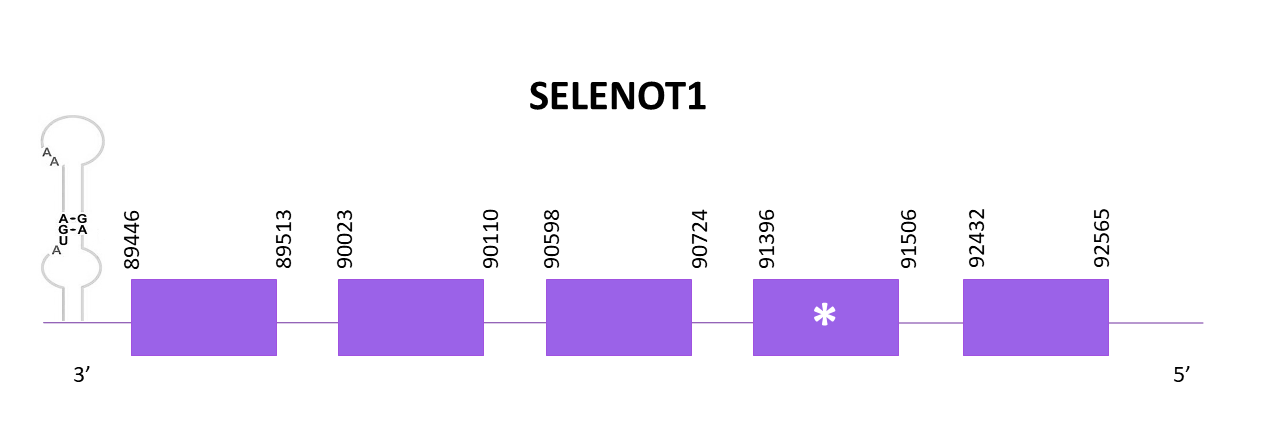
The predicted proteins obtained with Exonerate and Genewise are identical, both start with M but not with the first M of the reference sequence taking into consideration that there are 3 gaps in the start. The rest of the sequence is well aligned. Finally, using Seblastian there is no selenoprotein detected for this genomic region. Due to that, SECISearch3 is used and two SECIS structures are predicted in the 3'-UTR region.
SELENOT1b
When aligning the corresponding protein from Zebrafish (SPP00000651_2.0) to Helostoma temminkii’s genome, two scaffolds had significant hits. The first one is the one selected and it’s the same as SELENOT2 but located in different strands, and the second significant scaffold is the one assigned to the SELENOT1 protein.
The coding gene is located in the negative strand and goes from position 10880 to 11374 and according to Exonerate is composed of 3 exons. One selenocysteine is found as expected. One SECIS structure can be found, which concurs with SELENOT1b from Zebrafish.
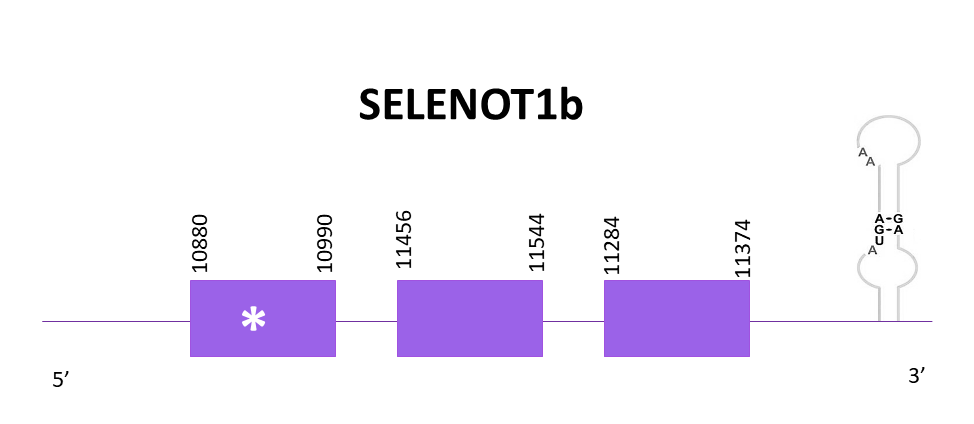
Both predictions obtained using Exonerate and Genewise are incomplete, they lack the start and the end, nevertheless, the Genewise one is better than the Exonerate one. Neither of the predictions (Exonerate, Genewise and Seblastian) include Met. The three predictions are pretty different and there is no criteria to choose which one is the best.
SELENOT2
In this case, two scaffolds showed significant hits when aligning the corresponding protein from Zebrafish (SPP00000650_2.0) to Helostoma temminkii’s genome, from these, OMLM01010512.1 showed the lowest E-value and the highest score and that’s why we selected this one. Also, the other scaffold corresponded to SELENOT1. As mentioned before, the selected scaffold also corresponds to the genetic coding region for SELENOT1b, but both proteins are located in different frames and strands.
The gene goes from position 7482 to 11565 in the positive strand and it consists of 6 exons, the protein contains a selenocysteine, located at the second exon. A SECIS structure can be found at the 3'-UTR region using Seblastian.
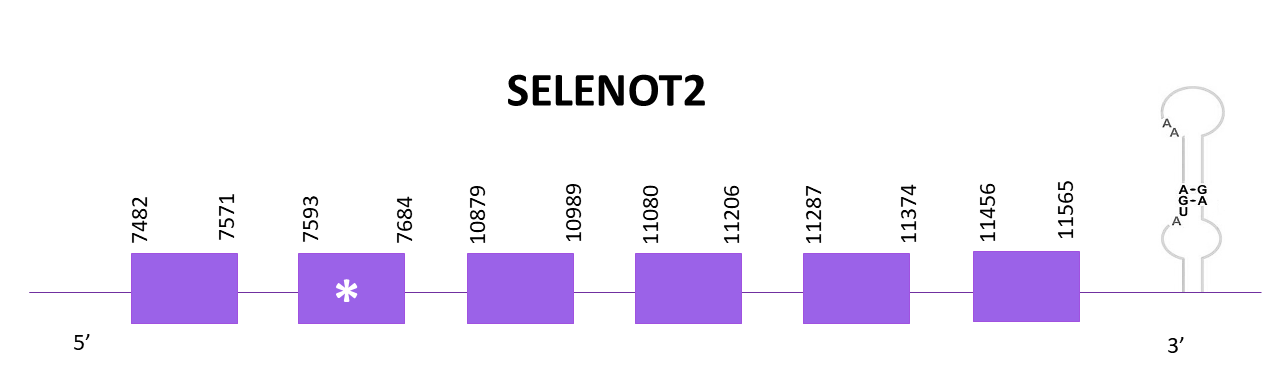
Both predictions obtained using Exonerate and Genewise are practically identical, presenting and initial methionine and only lacking 3 amino acids from the C-terminal region. In this case, the predicted protein from Seblastian does not have an initial methionine but it predicts the last 3 amino acids that were missing in the other predictions.
SELENOU is phylogenetically restricted in vertebrates such as chicken and fish, while other vertebrate members of this family, including mammals, contain cysteine-containing homologs.
SELENOU1a
Four scaffolds show significant hits when aligning with the corresponding protein from Zebrafish (SPP00000652_2.0) and Helostoma temminkii. The last one is the one assigned to SELENOU3 while the other 3 are equally good, that is why there are SELENOU1a, SELENOU1a.2, and SELENOU1a.3 (triplication of the gene).
The scaffold assigned to SELENO1a OMLM01011960.1 and according to Exonerate there are 4 exons and a selenocysteine conserved. The coding gene is located between 134886 and 135645 positions from the selected scaffold in its positive strand. One SECIS structure is predicted in the 3’ region like the Zebrafish SELENOU1a.
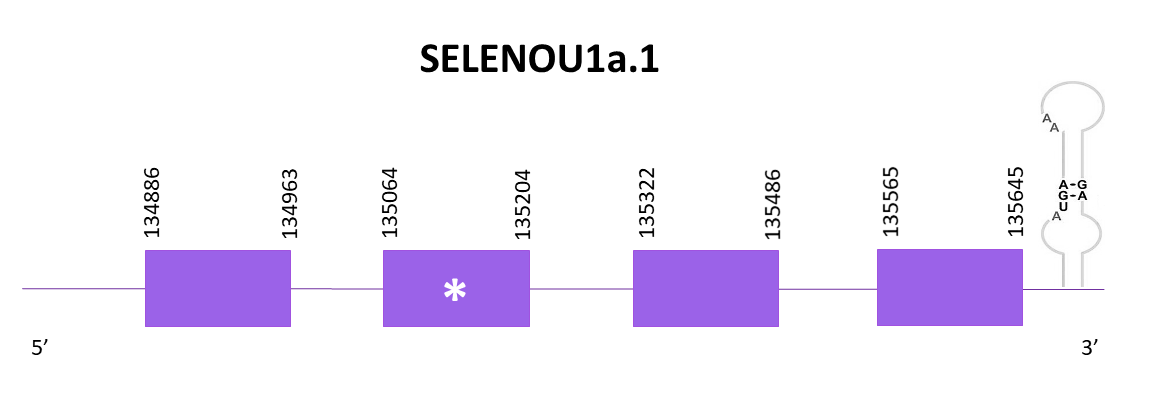
The predictions obtained with Genewise and Exonerate are similar, there are no loss of nucleotides in the final part of the sequence, however the start of the protein has gaps (more in Exonerate than in Genewise). When using Seblastian the prediction is larger than the one obtained with Genewise and it includes the initial Met.
SELENOU1a.2
As previously commented, the scaffold assigned to SELENO1a OMLM01011960.1 and according to Exonerate in this case there are 5 exons and also a selenocysteine conserved. The gene goes from 1988 to 4041 in the selected scaffold in its positive strand. Again, one SECIS structure is predicted in the 3’ region like the Zebrafish SELENOU1a.
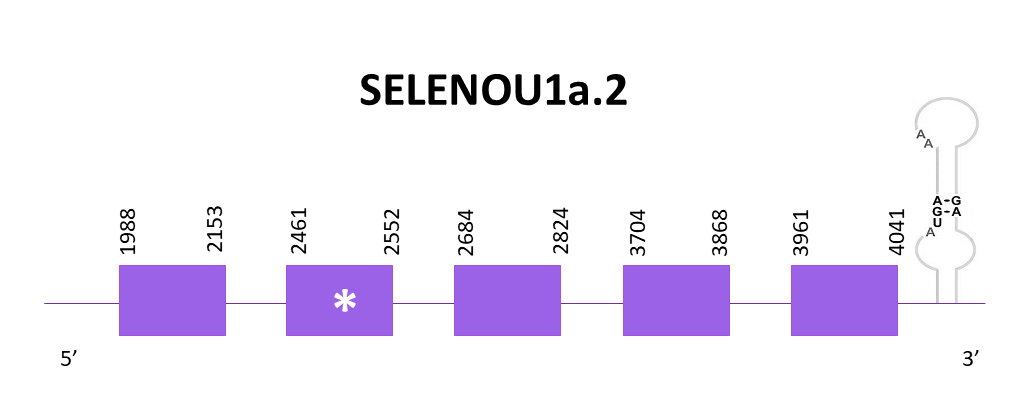
When using Genewise and Exonerate the prediction obtained are identical with only 7 amino acid lacking. The Seblastian prediction starts with Met and it starts after the Genewise and Exonerate predicted protein.
SELENOU1a.3
Again, the scaffold is OMLM01011960.1. With Exonerate, 4 exons are found and a selenocysteine is conserved as expected. The coding gene is located in the positive strand and goes from 48147 to 49223. In the 3'-UTR regions a SECIS candidate is found.
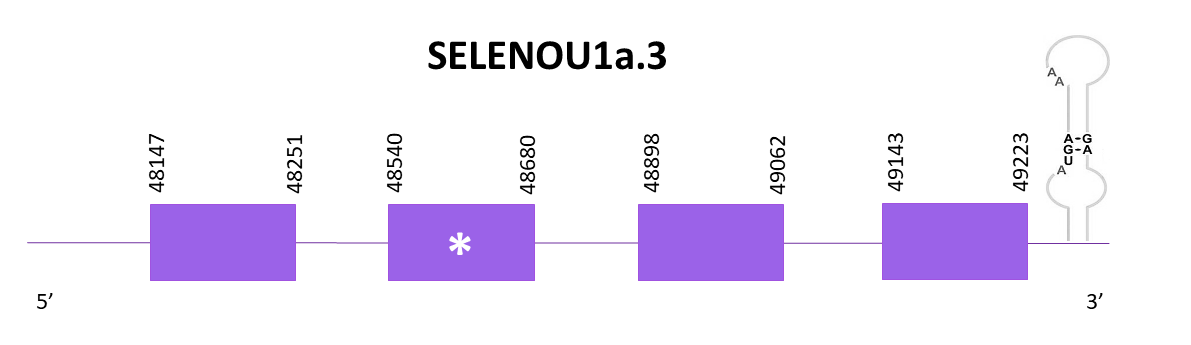
The protein prediction obtained with Genewise contains more amino acids in the start, so its better than the one obtained with Exonerate. The Seblastian prediction is not complete even if it is larger than the previous predictions.
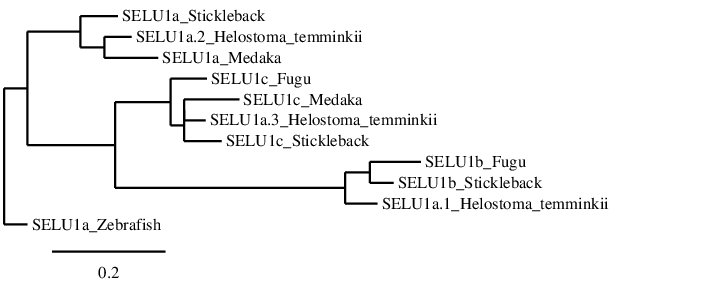
After analysing the selenoproteins found in Helostoma temminkii for SELENOU1a, we performed a phylogenetic tree to prove that the genomic regions were correctly selected for each protein and to validate that more than one duplication event for this protein took place in Helostoma temminkii’s genome.
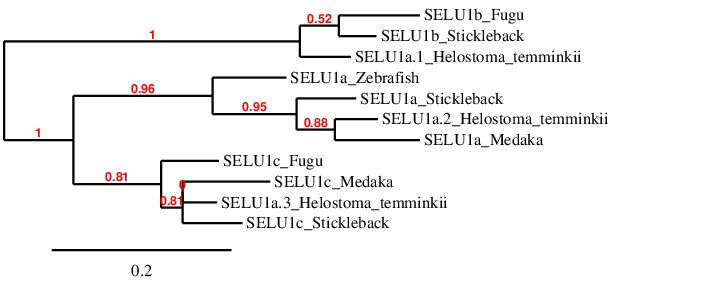
For this phylogenetic tree, SELENOU1s from several fishes (Fugu, Medaka, Stickleback and Zebrafish) were selected as a reference so as to correctly describe the proteins found in Helostoma temminkii. It has to be mentioned that there are three different SELENOU1 paralogs in total (SELENOU1a, SELENOU1b and SELENOU1c) and, from the reference fishes, only Stickleback presents all of them. As expected, each group of paralogs from the different species is grouped together and this fact helped us to infer that SELENOU1a.1 from Helostoma temminkii is in fact a homolog to SELENOU1b (and thus should be named like that), SELENOU1a.2 corresponds to SELENOU1a and SELENOU1a.3 corresponds to SELENOU1c. Consequently, Helostoma temminkii together with Stickleback are the only studied fishes that preserve the 3 proteins from the SELENOU1 subfamily.
In order to better understand what happened in the SELENOU1 subfamily through evolution and establish an hypothesis regarding the duplication and deletion events, we elaborated an schematic representation of the evolution of the bony fishes’ selenoproteome. The creation of a new selenoprotein (by duplication of an existing one) is indicated by its name in green. Loss is indicated in red.
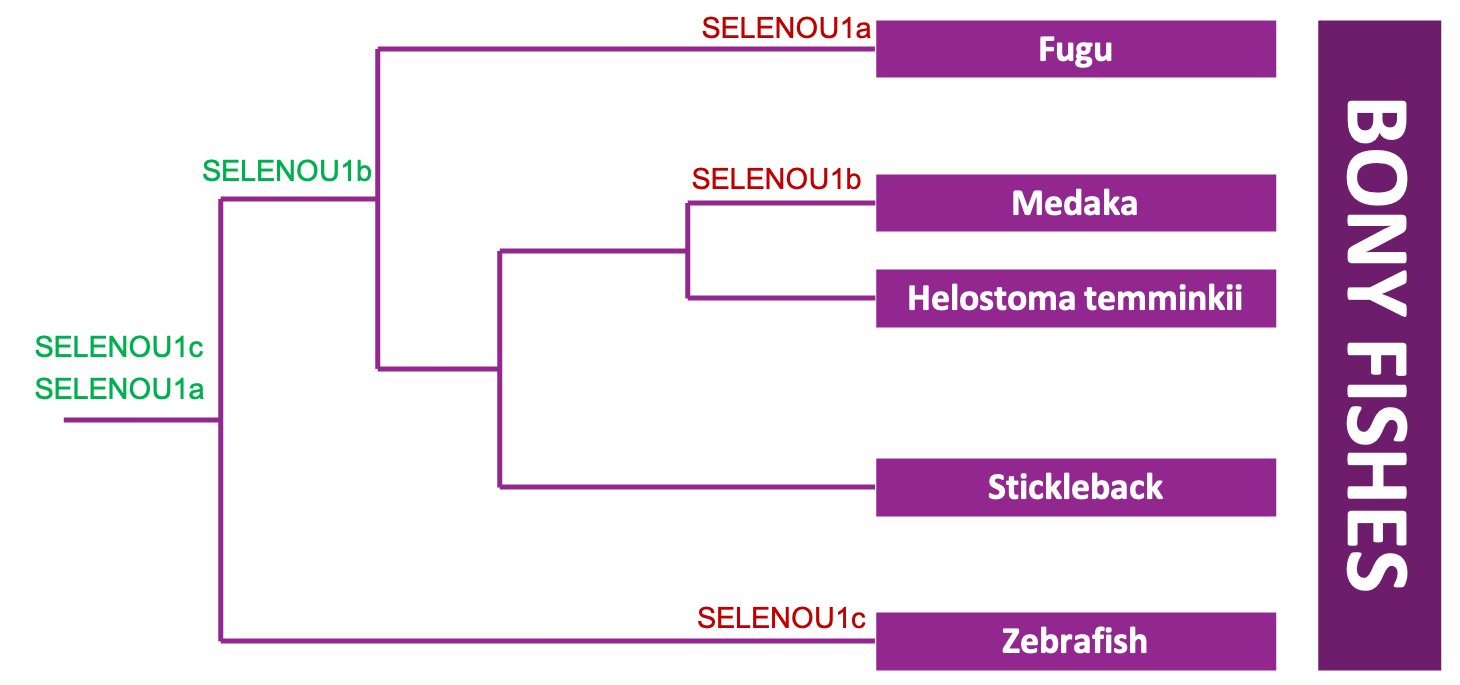
Our hypothesis is consistent with the idea that both SELENOU1a and SELENOU1c were present in the ancestral bony fish as a result of the the whole-genome duplication event that took place in bony fishes (Ts3R). Then, the first species to diverge was Zebrafish, which suffered a loss of SELENOU1c through its evolution since gene loss events are common and favoured after whole-genome duplications [46]. Regarding the other species, a second duplication event took place and a new protein was created (SELENOU1b). We believe that SELENOU1b is the result of a duplication from SELENOU1c, because they are more closely related to each other rather than to SELENOU1a as can be seen when rerooting the previously presented phylogenetic tree to SELENOU1a from Zebrafish, the most evolutionary different species from the studied ones.
The next species to diverge from the rest was Fugu, which lost SELENOU1a through its evolution. Finally, the three species left (Medaka, Stickleback and Helostoma temminkii) evolved and diverged from each other and only Medaka suffered a deletion of one of the three genes coding for SELENOU1s, losing SELENOU1b, the protein resulting from the second duplication. We want to point out that this is just our hypothesis and, to prove it, further studies should be carried out.
SELENOU2
In this case, only one scaffold showed significant hits when aligning the corresponding protein from Zebrafish (SPP00000655_2.0) to Helostoma temminkii’s genome, OMLM01014307.1.
The gene goes from position 122950 to 124461 in the positive strand and it consists of 6 exons, the protein does not contain a selenocysteine, thus being a cysteine homologue. No SECIS structure can be found for this protein.
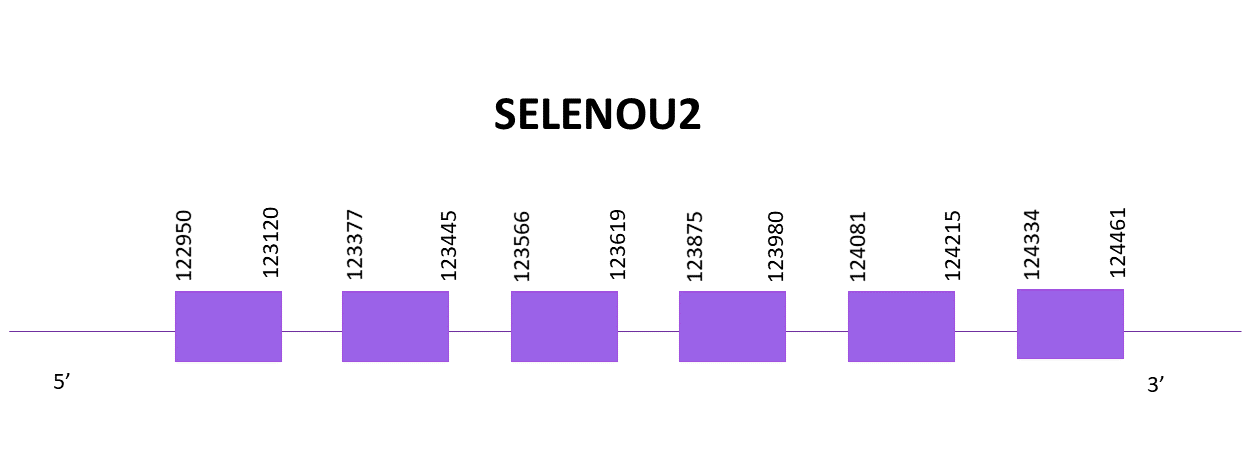
Both predictions obtained using Exonerate and Genewise are practically identical, only lacking 2 amino acids from the N-terminal region, the rest of the protein is well aligned.
SELENOU3
Just one scaffold had significant hits when comparing with Zebrafish’s protein (SPP00000656_2.0).
The coding gene for SELENOU3 is located in the negative strand between 2614 and 5560. When using Exonerate, 5 exons are predicted and a cysteine is found (like in the reference protein from Zebrafish).
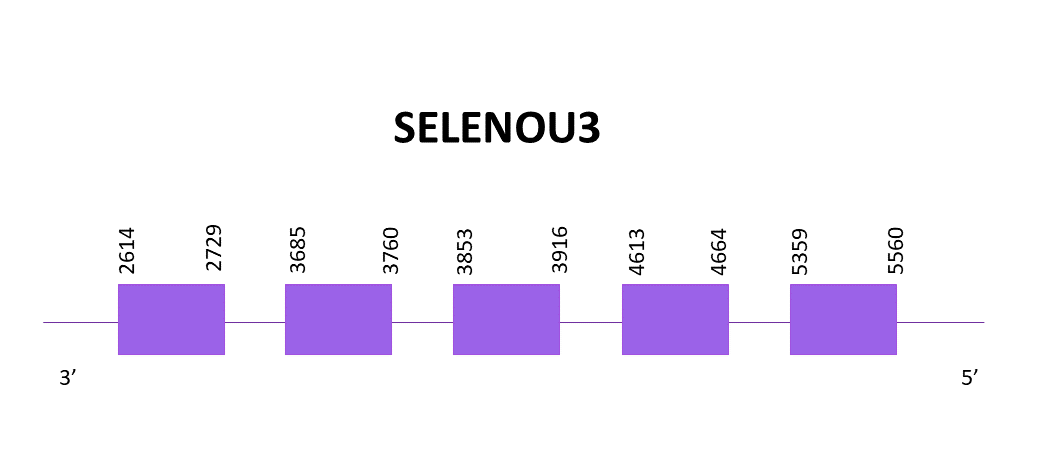
The predicted proteins obtained with Exonerate and Genewise are practically identical, missing some amino acids in the C-terminal region but the rest of the sequence is well aligned with the reference protein. Finally, using Seblastian there is no selenoprotein detected for this genomic region. Due to that, SECISearch3 is used and two SECIS structures are predicted in the 3'-UTR region. No SECIS can be found in Zebrafish for this protein and the SECIS predicted for Helostoma temminkii is located in the positive strand whereas our gene is located in the negative strand. For the mentioned reasons, we won’t consider the predicted SECIS as valid.
After analysing all the selenoproteins found in Helostoma temminkii from the SELENOU family, we performed a phylogenetic tree to prove that the genomic regions were correctly selected for each protein and to observe which proteins are more closely related within the SELENOU family.
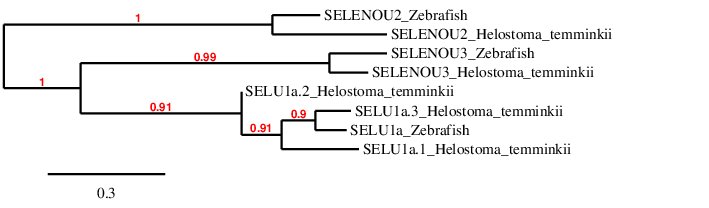
First of all, we can see that each protein from Helostoma temminkii’s SELENOU family is grouped together with the corresponding homolog from Zebrafish, proving that we have assigned the correct genomic region from our studied species to each protein. We can observe that SELENOU1 and SELENOU3 are more closely related within each other rather than with SELENOU2. However, we expected SELENO3 to be evolutionary closer to SELENOU2 since both of them are cysteine homologues.
SELENOW1 belongs to SelWTH family, which possesses a thioredoxin-like fold and a conserved CxxU motif, that suggests a redox function of this protein.
SELENOW_1
Only one scaffold has a significant hit, OMLM01020360.1, after comparing the Zebrafish protein (SPP00000658_2.0) with Helostoma temminkii’s genome.
Exonerate predicted 2 exons and the presence of a selenocysteine (as expected). The gene goes from 63508 to 63826 in the negative strand. A SECIS element is predicted in the 3'-UTR end like in the reference protein.
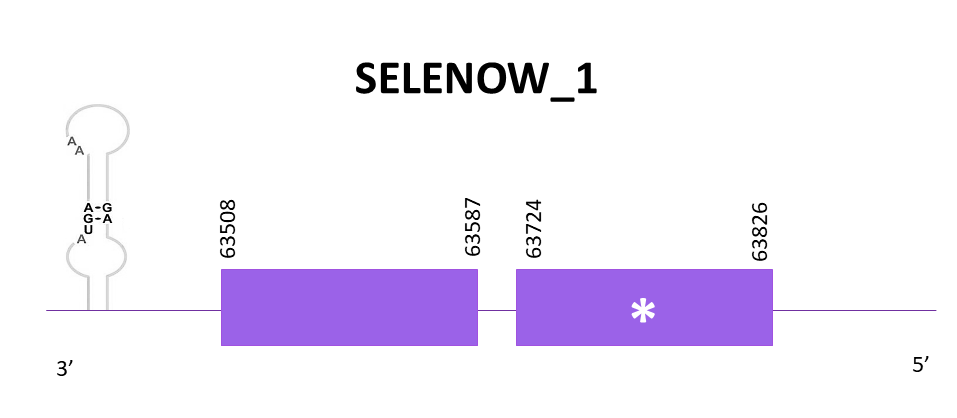
The prediction obtained with Genewise is better than the one obtained with Exonerate, taking into consideration the many gaps found in the second one. However, both predictions align few amino acids. Seblastian predicts the protein better (it includes the Met).
SELENOW_2
No significant hits were obtained for any scaffold when aligning the Helostoma temminkii’s genome with the corresponding protein from Zebrafish (SPP00000659_2.0). The E-values of the scaffolds obtained were higher than the threshold that we established for all the proteins. For this reason, we believe that SELENOW_2 is not present in Helostoma temminkii’s genome.
SELENOW_3
Like SELENOW_1, only one one scaffold shows a significant hit, so in this case the scaffold is the same as in SELENOW_1 when aligning Zebrafish (SPP00000657_2.0) with Helostoma temminkii’s genome. Nevertheless, when using Exonerate, 3 exons are predicted as well as a selenocysteine. The gene is located in the negative strand between 63508 and 64058. The first two exons correspond to the 2 exons predicted in SELENOW. One SECIS element is predicted using SECISearch3. Taking into account that the predicted gene for SELEOW_1 was located in the same genomic region and also the same strand, we decided to perform the same analysis but using Medaka’s SELENOW instead, since Medaka is more closely related to our species and better predictions might be obtained.
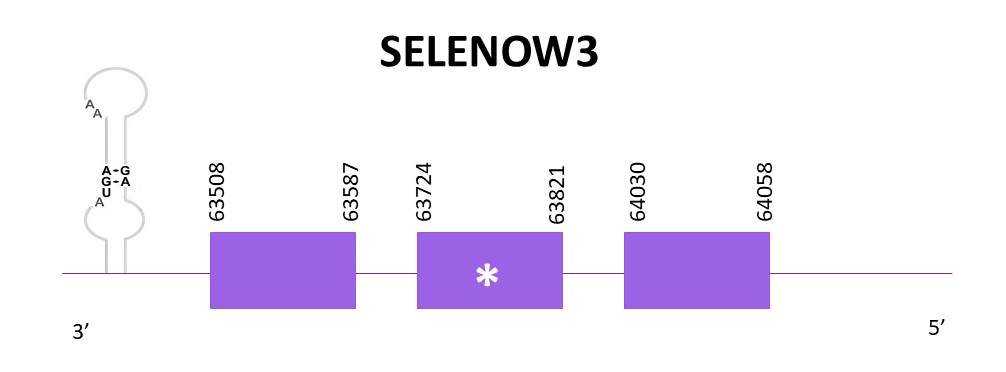
When using Exonerate and Genewise the protein predictions obtained are pretty different. The prediction of Exonerate lacks the last part of the protein even though it starts with Met. Genewise predicted protein lacks amino acids in the start. Sebastian predicts the complete protein.
SELENOW_Medaka
Again, the scaffold selected is OMLM01020360.1. In this case the references specie is not Zebrafish but Medaka for the reason previously explained. Exonerate predicted 3 exons and the presence of a selenocysteine (as expected). The gene goes from 63285 to 63820 in the negative strand. Also, a SECIS element is predicted in the 3'-UTR end like in the reference protein.
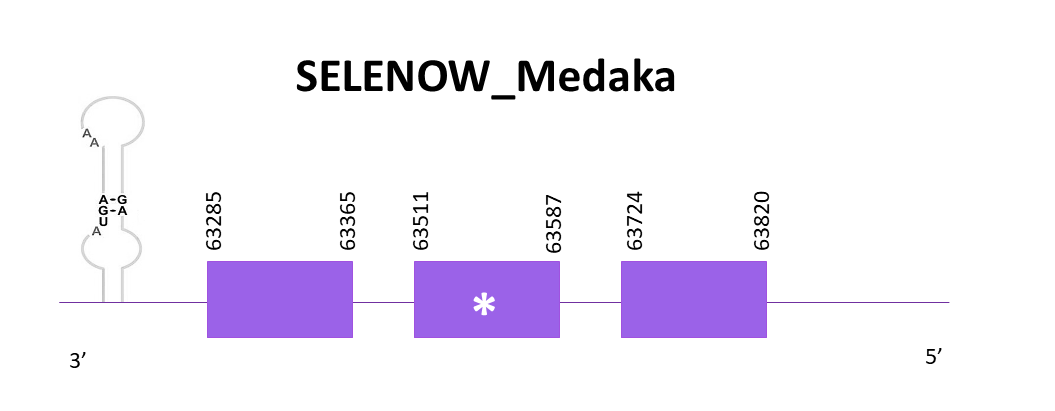
The prediction obtained with Exonerate is better than the one obtained with Genewise, due to the fact that it includes the first 3 amino acids present in the reference protein. The predictions are almost complete since they practically have the same length as the reference, a similar protein sequence is obtained using Seblastian although this one starts with a methionine. Comparing the predicted proteins with the ones obtained using Zebrafish as a reference, we can conclude that better results are obtained using a closer related species and that in Helostoma temminkii’s genome there is only one copy of SELENOW, like in Medaka’s genome.
In order to better understand our results, we attempted to perform a phylogenetic tree of SELENOW including the different proteins predicted and distinguishing duplications from copies in the exact same genomic region. However, since this protein is pretty short (less than 100 amino acids), its phylogenetic tree was not informative and its branches support values were very low. As a consequence, the tree is not shown. Our hypothesis is that both SELENOW_1, SELENOW_3 and SELENOW_Medaka predicted for Helostoma temminkii correspond to the same protein. Thus, only one copy of SELENOW is present in Helostoma temminkii’s genome.
SEPHS is in charge of the synthesis of selenophosphate, which is the active selenium donor. Two SEPHS paralogues are common in some eukaryotes, only SEPHS2 shows catalytic activity during selenophosphate synthesis. Nevertheless, SEPHS1 has been shown to play an essential role in regulating cellular physiology. In Danio rerio two homologs have been annotated: SEPHS1 and SEPHS2.
SEPHS1
Two different scaffolds showed significant hits when aligning the corresponding protein from Zebrafish (SPP00000629_2.0) to Helostoma temminkii’s genome. OMLM01014436.1 was the best candidate since it had the lowest E-value and highest score. The other scaffold corresponds to SEPHS2.
The gene coding for SEPHS1 is located between 28334 and 32815 positions from the selected scaffold in its negative strand. 9 exons are predicted for this protein using Exonerate and no selenocysteines are found in the protein, thus being a cysteine homologue. No SECIS structure is predicted, which concurs with the SEPHS1 from Zebrafish.
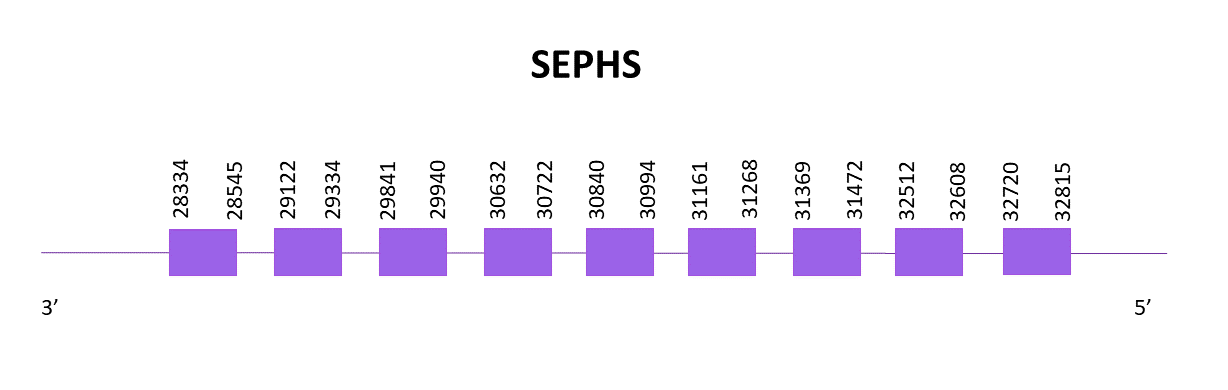
Proteins predicted using Exonerate and Genewise are identical and practically complete, starting with a methionine and predicting until the last amino acid of the reference protein. This indicates that SEPHS1 is very well conserved among different species and through evolution.
SEPHS2
Two scaffolds showed significant hits when aligning the corresponding protein from Zebrafish (SPP00000630_2.0) to Helostoma temminkii’s genome. OMLM01002683.1 was the selected scaffold since it had the lowest E-value and highest score. The other scaffold corresponds to SEPHS1.
The gene coding for SEPHS2 is located between 73046 and 78070 positions from the selected scaffold in its positive strand. 9 exons are predicted for this protein using Exonerate and a conserved selenocysteine is detected in the second exon. Despite that, no SECIS structure is predicted although it is present in both Zebrafish and Medaka.
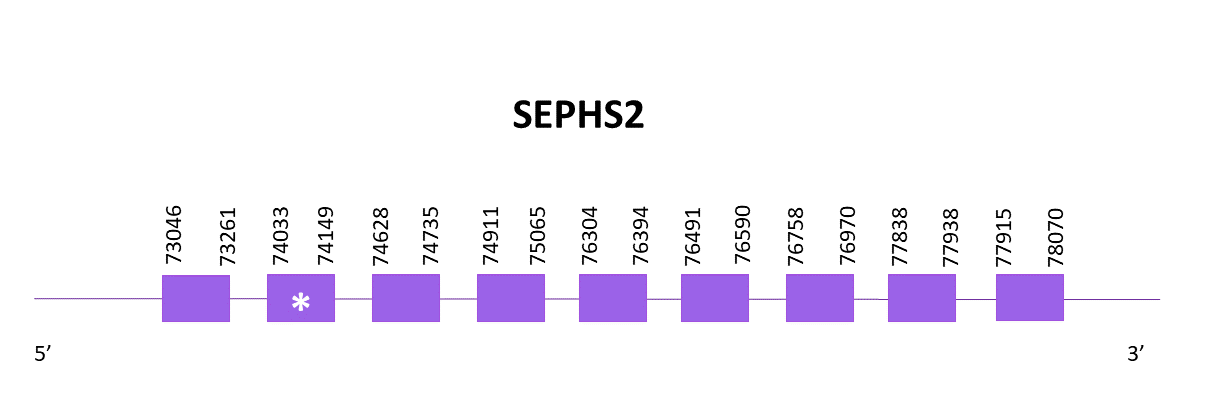
Proteins predicted using Exonerate and Genewise are similar, differences can be found in the central region of the protein were some gaps and unpredicted regions are located. Both predictions are pretty complete, they include the initial methionine and only lack a few amino acids in its central and C-terminal regions.
After analysing the proteins found in Helostoma temminkii from the SEPHS family, we performed a phylogenetic tree to prove that the genomic regions were correctly selected for each protein and to establish evolutionary relationships among species.
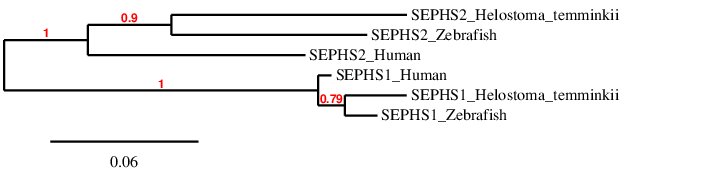
First of all, we can see that each protein from Helostoma temminkii’s SEPHS family is grouped together with the corresponding homolog from Zebrafish and Human, proving that we have assigned the correct genomic region from our studied species to each protein. Also, it can be seen that proteins predicted for Helostoma temminkii are more closely related to Zebrafish rather than to Human, which makes evolutionary sense. We can also point out that SEPHS1 homolog proteins are more similar between each other alternatively to SEPHS2, where the 3 homologous proteins have diverged more from each other and accumulated more differences (this can be inferred from the length of the branches in the phylogenetic tree, which is propotional to the evolutionary distance).
Thioredoxin reductases (TXNRDs) are oxidoreductases that, by combining with thioredoxin (Trx), comprise the major disulfide reduction system of the cell.
TXNRD2
Three different scaffolds showed significant hits when aligning the corresponding protein from Zebrafish (SPP00000660_2.0) to Helostoma temminkii’s genome, and, from these, the one selected (lowest E-value and highest score) was OMLM01003366.1. One of the other scaffolds was discarded since the alignment only represented a small portion of the whole protein and the identity values were very low. The other scaffold corresponded to TXNRD3.
The coding gene is located in the negative strand between 37010 and 53768. Using Exonerate, 15 exons were predicted for the protein and a conserved selenocysteine is found in the first exon.
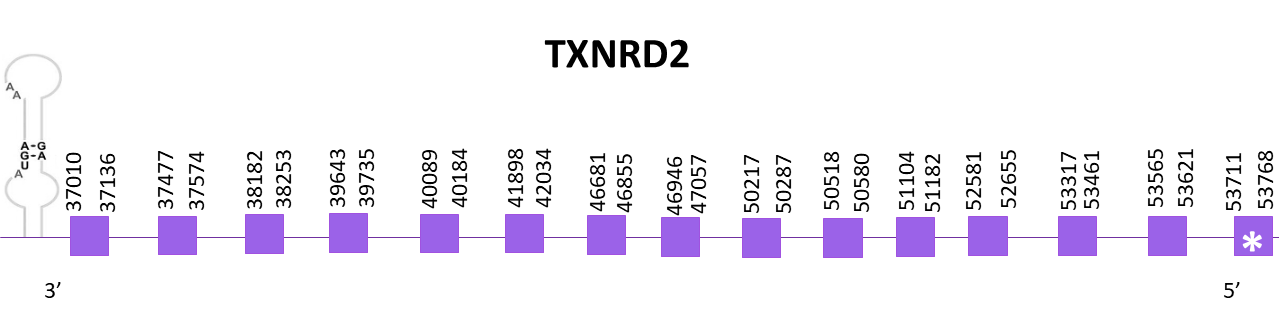
Protein predictions obtained using Exonerate and Genewise are practically identical and they are almost complete. However, neither the predicted proteins nor the reference protein from Zebrafish start with a methionine.
Finally, using SECISearc3, 2 SECIS structure are predicted although only one of them is considered as feasible since the other one is located in the positive strand and TXNRD2 is located in the negative strand.
TXNRD3
Different scaffolds showed significant hits when aligning the corresponding protein from Zebrafish (SPP00000661_2.0) to Helostoma temminkii’s genome, one of them being the genomic region corresponding to TXNRD2 and the rest being very short hits which only represent a small fraction of the protein and scores were also not good enough. The selected scaffold, OMLM01011900.1, had the highest score and the lowest E-values.
The coding gene is located in the negative strand between 58881 and 67244. Using Exonerate, 15 exons are predicted for the protein and a conserved selenocysteine is found at the first exon.
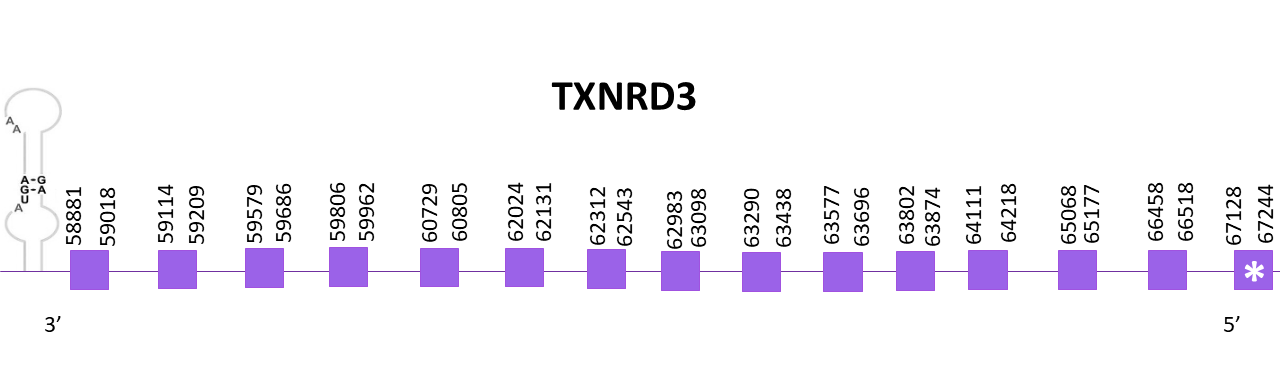
Similar predictions are obtained using Exonerate and Genewise. Both predictions present the methionine at the N-terminal end but lack some amino acids from the C-terminal region (being the Exonerate prediction the one lacking more amino acids compared to Genewise’s prediction).
Finally, using Seblastian a more extensive prediction of the protein for Helostoma temminkii is obtained, including the C-terminal end of the protein. A SECIS candidate is predicted in the 3'-UTR region using Seblastian.
After analysing the proteins found in Helostoma temminkii from the TXNRD family, we performed a phylogenetic tree to prove that the genomic regions were correctly selected for each protein and to establish evolutionary relationships among species.
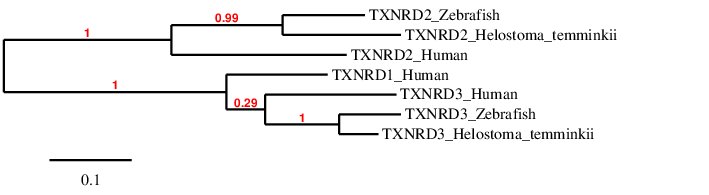
First of all, we can see that each protein from Helostoma temminkii’s TXNRD family is grouped together with the corresponding homolog from Zebrafish, proving that we have assigned the correct genomic region from our studied species to each protein. Also, it can be seen that proteins predicted for Helostoma temminkii are more closely related to Zebrafish rather than to Human, which makes evolutionary sense. We can also point out that TXNRD1 is only present in Human and this protein is more evolutionarily closer to TXRD3 rather than to TXNRD2, indicating that these two paralog proteins might be more similar.
In this case, only one scaffold showed significant hits when aligning the corresponding protein from Zebrafish (SPP00000662_2.0) to Helostoma temminkii’s genome, OMLM01010753.1.
The gene goes from position 13413 to 22703 in the positive strand and it consists of 7 exons, the protein does not contain a selenocysteine, thus being a cysteine homologue. No SECIS structure can be found for this protein like in the reference species.
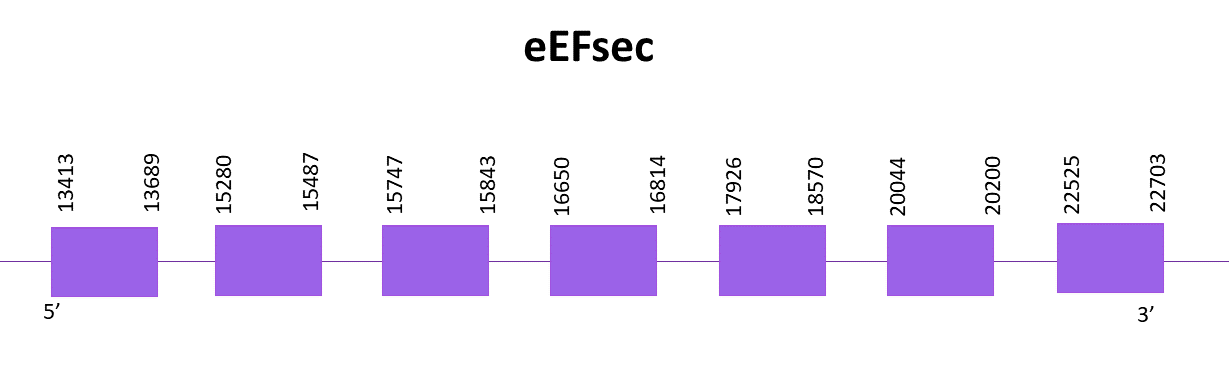
Both predictions obtained using Exonerate and Genewise are identical and almost complete, these only lack the first 5 amino acids. There is no selenoprotein detected for this genomic region with Seblastian nor SECISearch3.
When aligning the PSTK from Zebrafish (SPP00000663_2.0) and Helostoma temminkii’s genome, only one scaffold had a significant hit, OMLM01002072.1.
Using Exonerate one exon is found, as well as a Cys, so it is a Cys-containing protein. The gene goes from 67351 to 67494 in the positive strand of the selected scaffold.

The protein prediction obtained with Genewise is much better than the one obtained with Exonerate, knowing that the last one had gaps in the start but mostly in the end of the sequence. Neither one of the predictions is complete. There are no results with Seblastian.
PSTK_Medaka
PSTK is a protein machinery, and knowing that, the prediction should not be as bad as the one obtained when aligning with Zebrafish. Taking that into consideration, we align the protein with Medaka, a closer specie to Helostoma temminkii.
Like in the previous case, only one significant hit is obtained. When using Exonerate, in this case 5 exons are predicted, and also the Cys. No SECIS are found, with Seblastian nor SECISearch3, neither in Helostoma temminkii nor in Medaka.
The gene is located in the positive strand and goes from 67351 to 69442.
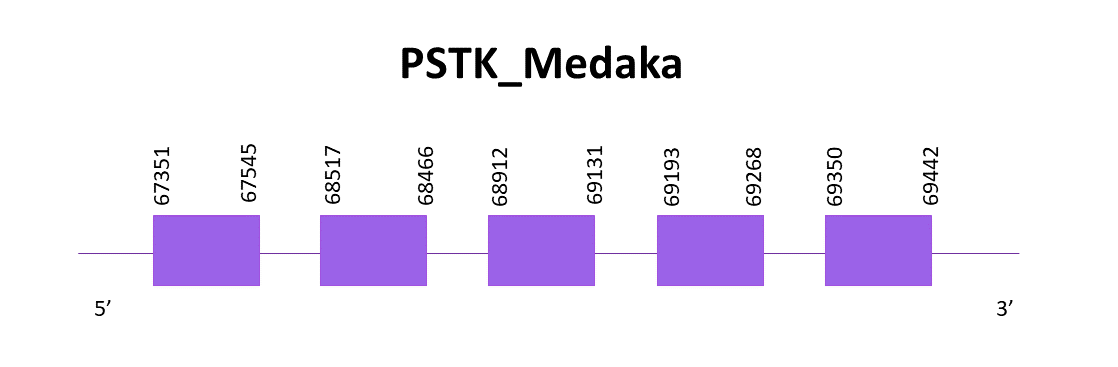
The predictions obtained with Genewise and Exonerate are identical and almost complete, only lacking the first amino acid (not a Met though). There are no results with Seblastian.
To conclude, in order to check whether the predicted PSTK protein is similar to its homologues in Zebrafish and Medaka we have performed a phylogenetic tree that is shown below:
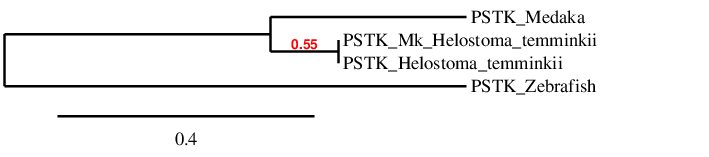
According to the phylogenetic tree, we were predicting the same protein from Helostoma temminkii using Medaka and Zebrafish as reference proteins (named PSTK_Mk_Helostoma_temminkii and PSTK_Helostoma_temminkii respectively), although a more extensive and complete prediction is obtained when using Medaka as a reference, as previously mentioned. Also, it can be seen that the protein predicted for Helostoma temminkii is more closely related to Medaka rather than to Zebrafish, which makes evolutionary sense since our species and Medaka diverged posterior to Zebrafish.
SBP2_1
When aligning with protein from Zebrafish (SPP00000627_2.0), two scaffolds show significant hits, the first one, OMLM01005182.1, with the best score and lowest E-value, is the one selected, while the second one is the one assigned to SBP2_2.
Using Exonerate 7 exons are found, as well as a Cys. The coding gene goes from 39571 to 42400 in the negative strand. One SECIS is predicted by SECISearch3 but it is not taken into consideration due to the fact that it is located in the positive strand and the gene is in the negative one.
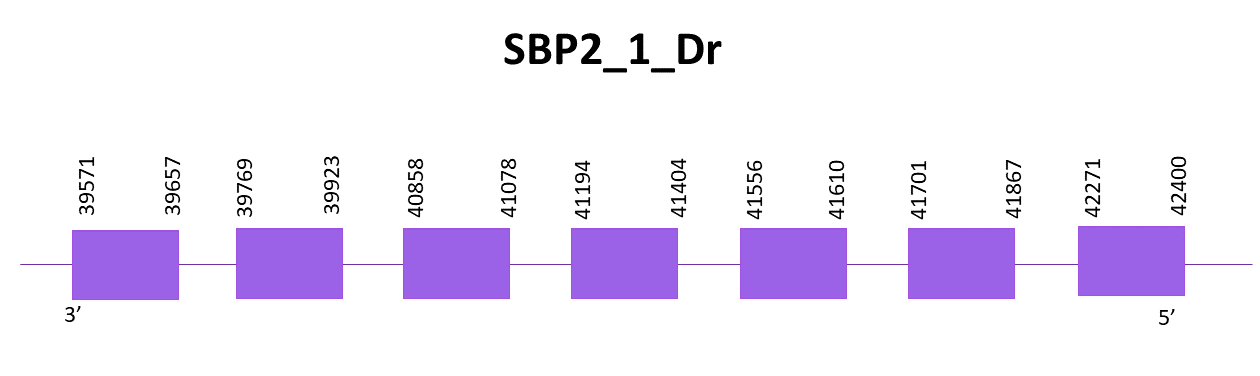
The predictions obtained with Genewise and Exonerate present some differences because of the central region that does not align correctly. The protein prediction obtained with Genewise shows more insertions than the one from Exonerate. There are no results with Seblastian.
SBP2_2
In this case, three scaffold showed significant hits when aligning the corresponding protein from Zebrafish (SPP00000627_2.0) to Helostoma temminkii’s genome, one corresponding to SBP2_1, another with a low identity, and the selected one OMLM01004801.1.
The gene goes from position 109 to 28447 in the negative strand and it consists of 14 exons, the protein does not contain a selenocysteine, thus being a cysteine homologue. One SECIS is predicted with SECISearch3 but it is found in the opposite strand, therefore we discard it.
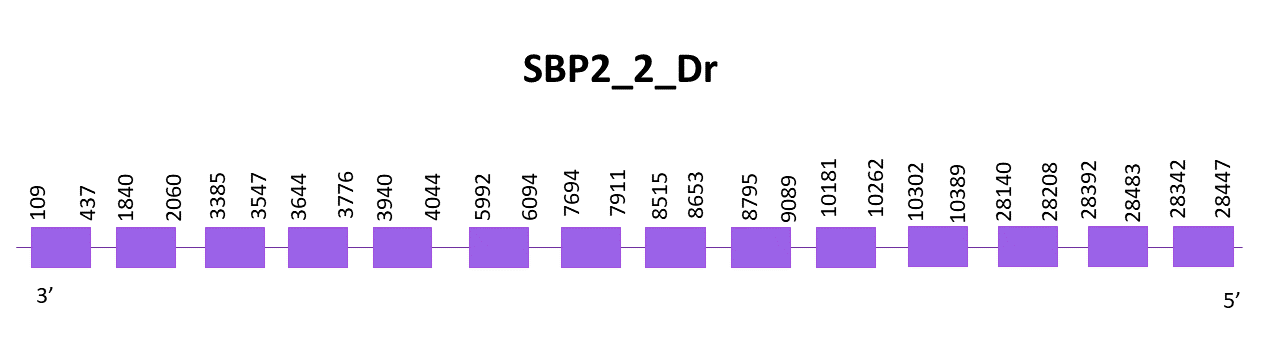
The predicted protein using Exonerate had lots of gaps, being better the prediction obtained with Genewise even if this one had many gaps. The prediction is incomplete, the last part is badly predicted and the initial part agrees better with the reference protein.
SBP2_2_Medaka
Taking into account that the predictions obtained when aligning with Zebrafish are not good enough, we also aligned Helostoma temminkii’s genome with Medaka’s SBP2 protein. The same scaffold as above is selected (OMLM01004801.1) despite this one is not the scaffold with the best score and E-value. Thus, we should also consider the latter scaffold (OMLM01001115.1).
Using Exonerate, 10 exons are predicted for OMLM01004801.1 scaffold, and, again, a Cys is found. The coding gene is located in the negative strand and goes from the 109 to the 4962 position.
Using Exonerate and Genewise, the predictions obtained are pretty similar, nevertheless, the one from Genewise had more gaps. In both cases the protein seems almost complete regarding the N-terminal region and central part of the protein, but it lacks several amino acids of the C-terminal end. Maybe the protein has lost the fragment or is truncated, the missing part being located in another scaffold.
As we have previously mentioned, another scaffold presented significant hits when aligning Helostoma temminkii’s genome with Medaka’s SBP2 protein. This alignment was pretty short and corresponded to the last part of the protein (C-terminal end) which was missing in the prediction made below with the other scaffold (OMLM01004801.1). We suspect that the gene coding for SBP2_2 protein is splitted between OMLM01004801.1 and OMLM01001115.1 scaffolds.
Using Exonerate, one exon is predicted for OMLM01001115.1 scaffold. The coding gene is located in the negative strand and goes from 49913 to the 50470 position.
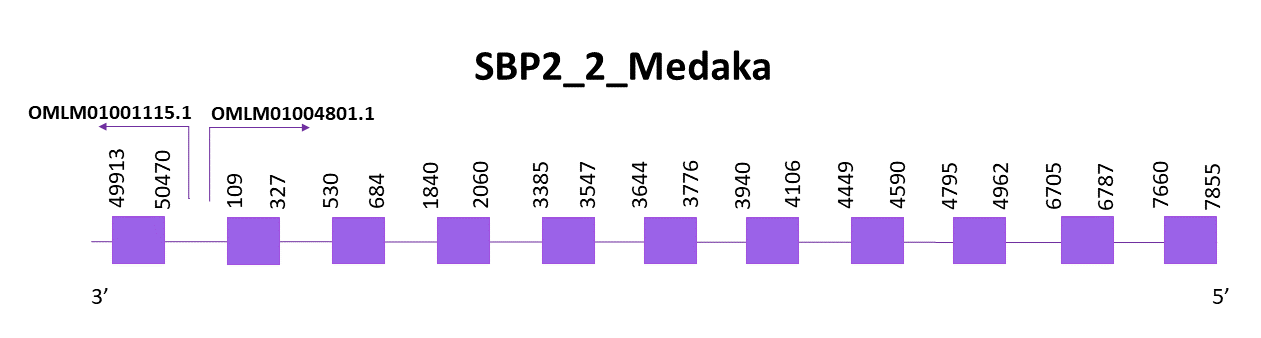
The proteins predicted using both Exonerate and Genewise are almost identical, they only align to the last part of the protein since, as it has already been said, this corresponds to the last part of the SBP2_2 protein.
After analysing the proteins found in Helostoma temminkii from the SBP2 family, we performed a phylogenetic tree to prove that the genomic regions were correctly selected for each protein and to establish evolutionary relationships among species.
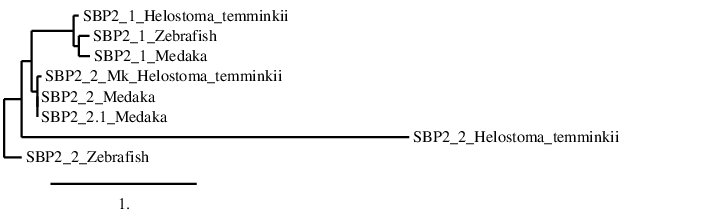
First of all, we can see that SBP2_1 from Helostoma temminkii is grouped together with the corresponding homolog from Zebrafish and Medaka, proving that we have assigned the correct genomic region from our studied species to this protein.
When it comes to SBP2_2, it can be observed that the Helostoma temminkii’s SBP2_2, predicted using Zebrafish as the reference protein (SBP2_2_Helostoma_temminkii), is not well aligned with the rest of SBP2_2 homologs; which could be explained by the fact that the prediction was not good enough because too many differences were accumulated between Zebrafish and Helostoma temminkii. On the other hand, Helostoma temminkii’s SBP2_2, predicted using Medaka as the reference protein (SBP2_2_Mk_Helostoma_temminki), is more closely related with the rest of SBP2_2 paralogs.
SECp43_1
5 scaffolds show significant hits when aligning Zebrafish (SPP00000664_2.0) and Helostoma temminkii. The selected one is OMLM01001339.1, which had the lowest E-value and highest score.
Exonerate predicted 4 exons and a Cys. The gene is located in the negative strand and goes from 8057 to 11136. No SECIS predicted with neither Seblastian nor SECISearch3.
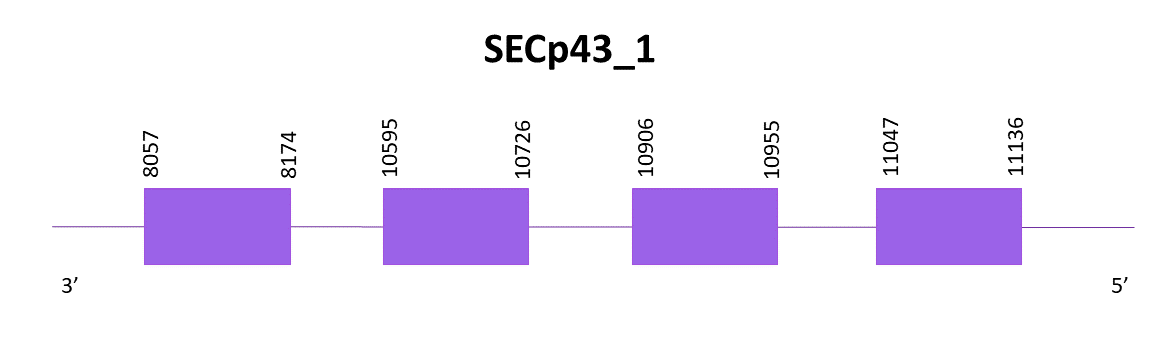
The predicted proteins with Exonerate and Genewise are different. The first one had a 44 amino acids gap, while the second one had a 9 amino acids gap.
SECp43_2
When aligning the Zebrafish protein (SPP00000665_2.0) and Helostoma temminkii, three scaffolds showed significant hits, and the one selected is OMLM01008182.1, taking into account that the other two were discarted because one had less than a 30% identity and the other was the one assigned to SECp43_1.
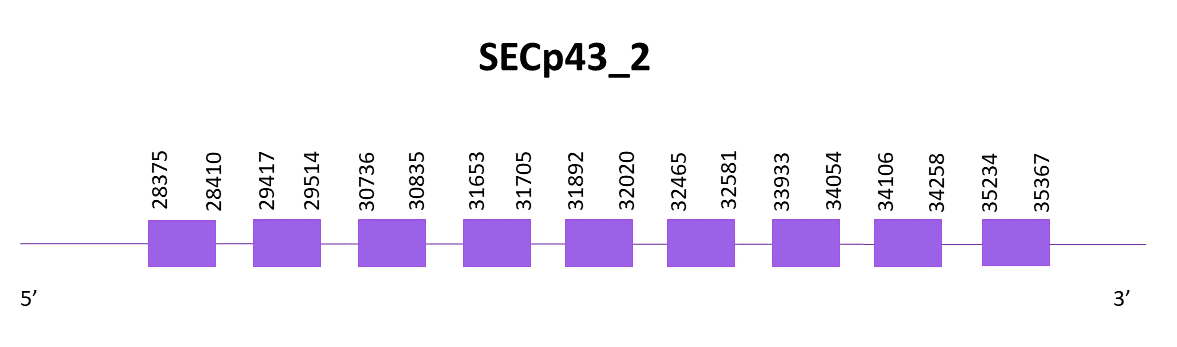
Using Exonerate, 9 exons were found, as well as the Cys. There are no SECIS predicted. Exonerate and Genewise protein predictions had the same length but the one from Exonerate had a better alignment and the prediction is complete.
After analysing the proteins found in Helostoma temminkii from the SECp43 family, we performed a phylogenetic tree to prove that the genomic regions were correctly selected for each protein and to establish evolutionary relationships among species.
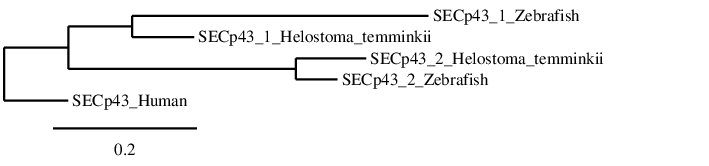
First of all, we can see that each protein from Helostoma temminkii’s SECp43 family is grouped together with the corresponding homolog from Zebrafish, proving that we have assigned the correct genomic region from our studied species to each protein. Also, it can be seen that proteins predicted for Helostoma temminkii are more closely related to Zebrafish rather than to Human, which makes evolutionary sense. Finally, it can be seen that the human SECp43 is separated from the fishes’ SECp43 and the duplication is only present in fishes, probably as a result of the whole-genome duplication event from bony fishes (Ts3R).
In this case, only one scaffold showed significant hits (high score and low E-value) when aligning the corresponding protein from Zebrafish (SPP00000631_2.0) to Helostoma temminkii’s genome, OMLM01012508.1.
The gene goes from position 13409 to 23568 in the negative strand and it consists of 11 exons, the protein does not contain a selenocysteine, thus being a cysteine homologue. No SECIS are predicted for this genomic region.

The predictions obtained with both Genewise and Exonerate are identical and lack some amino acids from the N-terminal region.