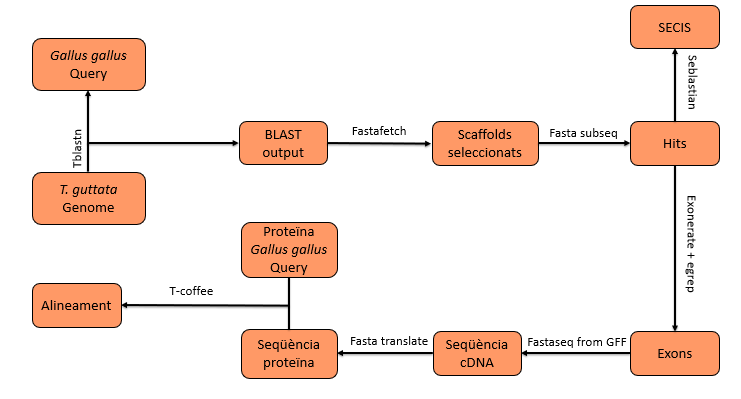
L’objectiu del nostre projecte és determinar i localitzar les selenoproteïnes del genoma de Taeniopygia guttata i també la maquinària necessària per a la seva síntesis i el seu funcionament. Per tal d’identificar les regions codificants per aquestes proteïnes, hem utilitzat el genoma de referència de Gallus gallus per la homologia que presenta.
Per tal d’obtenir les proteïnes del genoma de Gallus gallus, hem utilitzat una base de dades específica per a selenoproteïnes, SelenoDB. A partir d’aquí, hem obtingut les queries en format FASTA per a poder fer la comparació.
Manualment s’ha copiat cada seqüència amb l’editor de textos EMACS per tal d’eliminar alguns símbols (#,%,@) que es troben al final de la seqüència. Les selenocisteïnes es representen amb una “U”, però els programes que s’executen no reconeixen aquest caràcter. Per aquest motiu, hem substituït les “U” per el caràcter “X” que significa qualsevol aminoàcid. Una vegada realitzat aquest procediment, hem creat carpetes amb el nom de cada selenoproteïna i hem guardat el fitxer que conté la seqüència amb el nom de: selenoproteïna.fa.
El genoma de Taeniopygia guttata ens l’han proporcionat els professors de l’assignatura de Bioinformàtica. Es troba en el següent directori:
/cursos/20428/BI/genomes/2017/Taeniopygia_guttata/
Per realitzar la predicció, hem decidit automatitzar aquest procés per tal que fos més eficient. El nostre programa es troba en el següent enllaç.
El llenguatge que hem utilitzat és el “bash”.
Abans de fer córrer el nostre programa, hem carregat els mòduls necessaris per fer els alineaments. Per tenir permís de lectura, edició i execució del programa, hem utilitzat la següent comanda:
chmod u+x programa.sh
a) tBLASTn
El BLAST (Basic Local Alignment Search Tool) és un programa que serveix per comparar seqüències biològiques. És un dels programes més utilitzats en l’àmbit de la bioinformàtica. El BLAST utilitza un algorisme heurístic per trobar amb rapidesa les seqüències amb les que hi ha més homologia respecte la seqüència problema (query). Aquest programa et dona les significancies estadístiques d’aquestes similituds. Basant-nos amb aquestes dades, hem aplicat un llindar per quedar-nos amb les comparacions que són menys probable que passin per atzar. En aquest projecte, hem tingut en compte els e-value que estaven per sota del llindar de 1x10-10.
tblastn -query ${path_proteines}.fa -db ${path_db}/genome.fa -evalue 0.0000000001 -out ${path_proteines}.blast6 -outfmt 6
- Query: indica la seqüència problema (proteïna que estudiem del Gallus gallus
- db: és el genoma (base de dades) contra el qual volem fer el blast (Taeniopygia guttata)
- e-value: ens selecciona només els scaffolds que tenen un e-value menor que 1x10-10
- Out: és l’arxiu de sortida on volem que s’hi guardin els resultats obtinguts
- Outfmt 6: indica la forma amb la qual volem que ens doni el fitxer de sortida del blast
Després de realitzar el tblastn obtenim l’scaffold, que ens indica la localització de l’alineament en el genoma de Taeniopygia guttata; la identitat, que ens indica quant de similars són les dues seqüències; les posicions d’inici i de final, entre les quals trobem els hits i l’e-value, que ens indica el nombre de vegades que esperem trobar per atzar un alineament tant bo com el que hem obtingut.
Per continuar amb el procés, hem de triar els scaffolds que estudiarem tenint en compte la significança de l’e-value. Per aquest motiu, a dins del programa hem creat la variable “contador” que indica quin scaffold estem estudiant (el 1r, el 2n…). A partir d’aquest punt, el procés es realitza per a cada scaffold significatiu.
b) Fastafetch
El genoma ja ha estat indexat prèviament (genome.index). Per tant, ja podem passar a realitzar el fastafetch. Aquesta comanda ens permet extreure a partir d’un genoma que li donem (genome.fa), cada regió genòmica d’interès (scaffold) on hem predit els hits.
fastafetch ${path_db}/genome.fa ${path_db}/genome.index ${hit} > ${path_proteines}.${contador}.fastafetch.fa
c) Fastasubseq
Amb aquesta comanda podem extreure les regions específiques d'on es troba cada hit a dins de l’scaffold. Li hem de marcar unes coordenades: posició inicial i longitud.
Per assegurar-nos que la seqüència seleccionada conté el nostre gen d’interès, hem elongat 50.000 nucleòtids per sobre de l’extrem 5’ (upstream) i 50.000 nucleòtids per sota de l’extrem 3’ (downstream).
fastasubseq ${path_proteines}.$(contador)fastafetch.fa ${inici2} ${longitud} > ${path_proteines}.${contador}.subseq.fa
d) Predicció de gens: Exonerate
L’exonerate permet comparar la seqüència problema (en aminoàcids) amb el fitxer obtingut amb el fastasubseq (en nucleòtids) i permet obtenir les seqüències exòniques que codifiquen per les selenoproteïnes.
exonerate -m p2g --showtargetgff -q ${path_proteines}.fa -t ${path_proteines}.${contador}.subseq.fa
- m: ens indica el tipus d’alineament, en aquest cas “protein to gene” p2g
- showtargetgff: indica que el fitxer de sortida serà en format gff
- q: és la query
- t: és la regió resultant del fastasubseq
Després de realitzar aquesta comanda, tal com hem comentat anteriorment, ens hem de quedar únicament amb els exons (cDNA). Amb l’egrep -w exon podem seleccionar les files del fitxer GFF que contingui la paraula “exon”.
exonerate -m p2g --showtargetgff -q ${path_proteines}.fa -t ${path_proteines}.${contador}.subseq.fa | egrep -w exon > ${path_proteines}.${contador}.exonerate.gff
FastaseqfromGFF
El programa exonerte ens ha creat la seqüència de cDNA en format GFF. Nosaltres la volem tenir en format FASTA. Per fer-ho, utilitzem la següent comanda:
fastaseqfromGFF.pl ${path_proteines}.$(contador).subseq.fa ${path_proteines}.$(contador).exonerate.gff > ${path_proteines}.${contador}.nuc.fa
Fastatranslate
Ara, tenim la seqüència de nucleòtids del cDNA i la volem traduir a aminoàcids per obtenir la seqüència proteica del genoma de Taeniopygia guttata.
fastatranslate -F 1 ${path_proteines}.${contador}.nuc.fa > ${path_proteines}.${contador}.pred.aa.fa
e) Alineament final: T-Coffee
Amb el programa T-Coffee (Tree-based Consistency Objective Function for alignment Evaluation) comparem la seqüència problema (la obtinguda a partir de SelenoDB) amb la proteïna que hem predit de Taeniopygia guttata. El fitxer resultant ens mostra la homologia entre les dues seqüències i els aminoàcids de l’alineament més probable. Ens permet fer l’alineament global i informar-nos de la seva homologia.
De forma manual hem seleccionat els scaffolds que tenen un alineament més bo per poder fer la predicció de SECIS i estudiar-los amb profunditat.
t_coffee ${path_proteines}.${contador}.pred.aa.fa ${path_proteines}.fa > ${path_proteines}.${contador}.tcoffee
La predicció dels elements SECIS és important perquè són els elements responsables de la síntesis del codó de la selenocisteïna a partir del codó UGA. Aquest és el motiu pel qual, utilitzem els elements SECIS per confirmar la predicció de selenoproteïnes.
Per predir els SECIS hem utilitzat el programa Seblastian. També hem utilitzat el programa SECISearch3/Seblastian
Una vegada hem obtingut els gens de Taeniopygia guttata, hem utilitzat el software mencionat anteriorment per predir els elements SECIS a la regió genòmica de cada proteïna. Com a input hem donat el fitxer “fastasubseq” i hem donat la opció al programa que ens predigui tots els SECIS predits, d’aquesta forma hem pogut seleccionar amb el nostre criteri amb quin SECIS ens quedem.
Criteris per seleccionar els elements SECIS:
- L’element SECIS i el gen han d’estar al mateix marc de lectura (“frame”).
- L’element SECIS ha d’estar localitzat a la regió 3'UTR.
- Si un element SECIS està a una distància de 800 o més nucleòtids del UGA, l’hem descartat.


