
METHODOLOGY
The main objective of this project is to predict and annotate Oncorhynchus mykiss's selenoproteins, the selenoprotein translational machinery and the proteins related to selenium metabolism. We have compared the genome of Oncorhynchus mykiss with Danio rerio's (also known as zebrafish), another fish specie that we have considered phylogenetically suitable and completely annotated.
Obtention of Oncorhynchus mykiss' genome
The Oncorhynchus mykiss' genome (genome.fa) was provided by our Bioinformatics teachers, it was available in the following directory:
Obtention of reference genome
In order to predict Oncorhynchus mykiss's proteins, we have chosen the zebrafish as reference. We have considered zebrafish as our reference due to their phylogenetic distances, and also because its selenoproteins are well described and annotated since its importance as a research model. All data was extracted form the selenoprotein database SelenoDB.
Some Danio rerio' protein families did not present the subfamily specificated, annotated as "NONE". We renamed this proteins with the name of the family followed by a number. For example, the Selenoprotein W (SELENOW) family presents three proteins annotated as "NONE", which were renamed as SELENOW_1, SELENOW_2 and SELENOW_3.
Prediction process
We created two Perl programmes in order to automatically generate all the relevant files for the protein prediction. Data analysis and final predictions were performed afterwards.
Perl programmes
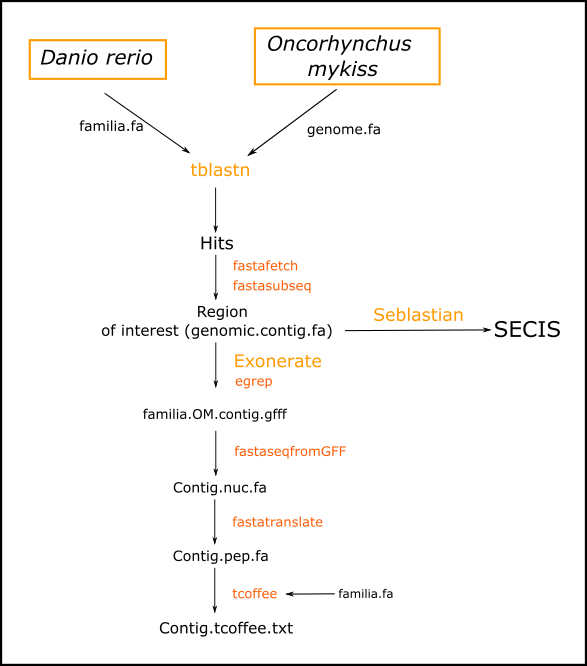
Both perl programs were based in the scheme shown in Figure 1 and are available in the following links:
Parugrama.pl is able to generate all the necessary files for protein prediction from a query (SUBFAMILIA). Despite that, this program presents some limitations because of its automated grade. Its main limitations were related to determining genomic positions of some hits, so we used Programet.pl since it allows to analyze each tblastn hit independently and introduce manually the starting and ending position of each hit
.BLAST
To compare each query to Ocorhynchus mykiss's genome we performed a BLAST (basic local alignment search tool), an algorithm used for comparing DNA sequences and selecting significant hits for further analysis. We concretely displayed a tblastn, a function that aligns the protein sequence of the query with the genome of Oncorhynchus mykiss.
To assess the significance of each hit we took into account the E-value. It describes how many times you can expect to find an alignment as good as that one in your database only by chance, so that lower E-values correspond to higher significances. For this reason we took the hits with E-values lower than 0.0001 for further analysis.
We generated an output document containing the following elements for each hit:
- Contig: corresponds to the genome fragment in which the hit was found.
- Start: Initial position of the hit.
- End: Ending position of the hit.
- E-value: Measure of the significance of the hit.
Substitution of U's for X's
Protein sequences extracted from SelenoDB anotate selenocysteines as U'. However, when running the programmes detailed right after, an error showed up since these U' were not recognized as any amino acid. Hence, all tblastn output files were processed with the following programme in order to convert all the selenocysteines (U) into X.
FASTA FETCH
We used this command to extract each scaffold from the hits across the given genome, corresponding to the region that contains the hit.
FASTA SUBSEQ
Once we have selected the scaffold, we extracted the concrete region of each hit (region of interest) by using the fasta subseq command. The main problem of this procedure is that the hit only represents coding sequences of the genome. We solved this by extending the search 50.000 nucleotides in both 5' and 3' directions, for not missing a part of the sequence because of the introns.
There were some cases where was impossible to extend 50.000 nucleotide in both directions (e.g. short contigs or genes located near the start of the chromosome). In these cases, we used Programet.pl to introduce the number of nucleotides that the extension will make.
EXONERATE
Once we have extracted our region of interest, we used exonerate to extract the different exons of the predicted gene found. The egrep function fused these exons into the same file.
FASTA SEQ FROM GFF
This command is used to generate a file containing the cDNA of the predicted protein based on the results of the exonerate.
FASTA TRANSLATE
This program translate the cDNA into a protein sequence. The output file obtained corresponds to the primary structure of our predicted protein.
Substitution of *'s for X's
The fastatranslate output file converts all predicted selenocysteines (initially anotated as X) into "*", which causes an error when running T-coffee. In order to avoid this, just after fastatranslate finishes, the following code line transformes all "*" found in the output file into X's:
T_COFFEE (Tree-based Consistency Objective Function for alignment Evaluation)
This programme was used to make a global alignment between our predicted protein sequence and the query protein from Danio rerio.
SECIS and SEBLASTIAN
As said before (see Introduction), SECIS are necessary elements in selenoporteins mRNA in order to present Sec in their sequences. Seblastian is the computational approach that we used to identify SECIS element in our predicted sequences. If no SECIS elements were predicted, we also used SECISearch3 in order to corroborate these findings. .
Phylogenetic tree
After all proteins in Oncorhynchus mykiss were found, we made a phylogenetic tree for each protein family using a tool available at phylogeny.fr. For each family, the input file had the predicted protein sequence in Oncorhynchus mykiss and the protein sequence of its homolog in Zebrafish and in human.
