Methods
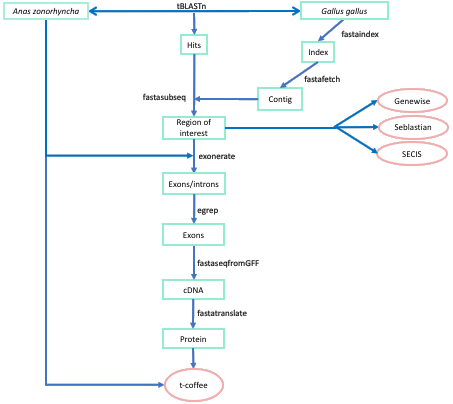
The aim of our project is to find, identify and annotate all the Anas Zonorhyncha's selenoproteins. The methods we have used to achieve this objective are mainly based on the software NCBI BLAST. We have compared the genome of our specie of interest (Anas Zonorhyncha) with the one of the chicken (Gallus gallus), as it is a phylogenetically close specie whose selenoproteins are annotated in the database SelenoDB [28,29]. In addition, we have used the human (Homo sapiens) selenoproteome to compare those selenoproteins that were not present in Gallus Gallus as well as the ones not well-annotated in the data base.
Queries acquisition
In order to select the queries that are going to be compared with Anas Zonorhyncha's genome, we have chosen Gallus gallus as a reference, as explained before. Its selenoproteome was provided by the database SelenoDB (a selenoprotein specialized database). However, we have found some problems during this process, as this selenoproteome can be found only (he quitado el just de antes) in SelenoDB 2.0 and not in SelenoDB 1.0, which means that it has been annotated automatically and has never been checked manually. Thus, there is some risk of selenoproteins not being well annotated. For this reason, why we have had to check if they all start with a methionine residue, because if it is otherwise it might mean that the whole protein has not been correctly annotated.
In order to determine whether the protein is well annotated, we have compared the SelenoDB transcripts which do not start with methionine with the Ensembl [30] transcript of the same protein. For these chicken proteins with a clear annotation mistake, we have repeated the whole process using SELENO DB 1.0 human protein transcripts and the Ensembl protein transcripts as queries, in order to predict a more accurate protein.
Manual prediction of selenoproteins
Genome selection
We were provided with the Anas Zonorchyncha's genome. It could be obtained from the following path:
| /cursos/20428/BI/genomes/2017/Anas_zonorchyncha/genome.fa. |
tBlastn
We have used BLAST to compare each query selected with the Anas zonorhyncha genome. Each Gallus gallus selenoprotein has been copied from SelenoDB and saved as a fasta file (selenoproteinname.fa) after erasing special characters (such as @ or #) in order to avoid reading errors during the execution of the program. The command we used to execute the Blast is the following:
| blastn -query selenoproteinname.fa -db /cursos/20428/BI/genomes/2017/Anas_zonorhyncha/genome.fa -out tblastn_selenoproteinname.fa |
| tblastn -query selenoproteinname.fa -db /cursos/20428/BI/genomes/2017/Anas_zonorhyncha/genome.fa -outfmt 7 -out tblastn2_selenoproteinname.fa |
There will be two output files. The first one is a fasta file with the different hits obtained from the comparison of each query protein from Gallus gallus in our Anas Zonorhyncha genome, as well as other relevant information such as the aligned sequences, the scaffold it is located, the E value and the value of both score and identity. This information will be used to determine the best candidates to further analyse them.
Fasta fetch
We execute a fastafetch command to extract from the Anas Zonorhyncha genome the genomic region that potentially contains the gene we are looking for. In order to do so, we have design a pre fasta fetch program that analyses the information obtained from the blast search and automatically select the best candidates as the ones that accomplish the following conditions: Its percentage of identity has to be >75%, its alignment lenght >25 and finally its E value <0.000001.
Using this information and the indexed genome we have been provided, that make it easier for the fasta subseq to find sequences in the whole genome, we executed the fastafetch and we obtained one fasta file (contigname.fa) for each scaffold that contains a valid hit. The command executed is the following:
| fastafetch /cursos/20428/BI/genomes/2017/Anas_zonorhyncha/genome.fa /cursos/20428/BI/genomes/2017/Anas_zonorhyncha/genome.index 'contigname' > contigname.fa |
We also need to consider that the different proteins of a family cannot have the same chosen contig.
Fasta subseq
The following step is to execute a fastasubseq command that extracts from the contig file just the region we are interested in, the one aligned with the query in the blast performed. The program requires the determination of the starting point and the length of the sequence to be extracted. To do so, we have designed a program that automatically obtains these numbers and provides them to the fastasubseq program.
This program's output is a file named 'numeros_proteinname', which contains the following information: in the first column there are increasing numbers for each contig, which indicate to which hit from the TBLASTN output each line refers to in case there is more than one hit for a contig; in the second column we find the name of the contig; the third column indicates with a 1 or a 0 if the hit is located in the forward or reverse strand, with 1 meaning that the sequence is in the forward strand and 0 meaning that it is in the reverse strand; in the fourth column there is the start position and finally, in the last column, there is the length of the alignment. The program also extends 50.000 basepairs in both directions, 5' and 3', of the aligned sequence (changing the initial position and the length), as we wanted to ensure that the complete gene was extracted. The program assigns automatically the first position of the contig to the start of the fastasubseq in case the extension on the 5' direction surpasses its start, or limits the length to the last nucleotide of the contig in case the extension on the 3' direction surpasses its end.
Once this program is executed, fastasubseq is ready to use the output obtained. The output of the fastasubseq will be saved with the name 'fastasubseq_contigname_hitnumber.fa'. The command that executes the fastasubseq is the following:
| fastasubseq 'contigname.fa' initialposition length > fastasubseq_contigname_hitnumber.fa |
Exonerate
The program exonerate uses the query protein file in order to do an exon prediction from the fastasubseq output to obtain a new file containing just the exonic sequence of the genome of Anas zonorhyncha that codifies for the protein.
We have added two extra commands, a sed one to replace every U for a * in the sequence, and the egrep -w command in order to just select the lines containing an exon and we have created an output file for each of them, named 'exon_fastasubseq_contigname_hitnumber.fa.gff'. The command that executes the exonerate is the following one:
| exonerate -m p2g --showtargetgff -q 'selenoproteinname.fa' -t 'fastasubseq_contigname_hitnumber.fa' |
Where the meaning of each command is:
- -m p2g: compares the protein with the genome.
- --showtargetgff: allows us to get all the information about the coordinates in a GFF file format.
- -q: shows that the file we are using is a FASTA format.
-t: is the target, the sequence we obtain from doing the fastasubseq command.
Fastaseq from GFF
Fastaseqfromgff is a program that converts the gff file obtained in the exonerate into a pl file (preprot_fastasubseq_contigname_contignumber.pl) that allows the execution of the following step, the fastatranslate. The command is the following one:
| fastaseqfromGFF.pl 'fastasubseq_contigname_hitnumber.fa' 'exon_fastasubseq_contigname_hitnumber.fa.gff' |
Fasta translate
This program translates the cDNA sequence of the exons to an amino acid sequence. In this step, we obtained the predicted Anas zonorhyncha's protein and we saved it in a fasta file (prot_fastasubseq_contigname_hitnumber.fa). The command executed is:
| fastatranslate -f preprot_fastasubseq_contigname_hitnumber.pl -F 1 > prot_fastasubseq_contigname_hitnumber.fa |
T-coffee
This programs compares the query sequence obtained from SelenoDB with the sequence of the Anas zonorhyncha's protein that has been predicted. The output is a global alignment of both sequences, together with the information necessary to determine the homology between them.
We have added a sed command, previous to the TCoffee command, in order to undo the replacement of the TGA codon for * that occurs in the fastatranslate command. The output obtained was saved in a text file by the name of tcoffee_fastasubseq_contigname_hitnumber.fa.txt. It is executed with this command:
| t_coffee selenoproteinname.fa prot_fastasubseq_contigname_hitnumber.fa |
SECIS elements and selenoprotein predictions by Seblastian
Seblastian [31] is an online tool that allows to find SECIS elements in a given sequence, and then performs a blastx on the sequence upstream of the SECIS against a comprehensive database (which includes selenoproteins), in order to find a selenoprotein that corresponds to the found SECIS element. However, the fact that Seblastian is able to predict a selenoprotein does not ensure that such protein is found in the genome of Anas zonorhyncha, although it does strengthen the hypothesis that the genome contains such selenoprotein. On the other hand, the fact that Seblastian cannot predict a selenoprotein does not imply that such protein is not found in the genome, as it might not be in any database.
Automatic prediction of selenoproteins
An automatic program for the search and analysis of selenoproteins in the genome of Anas zonorhyncha has been created. This program is able to run all the steps (from BLAST to TCoffee) automatically, and it only needs someone to manually specify in its code which proteins are we looking for, which must be placed in a specific path. The program can be modified to use different queries from SelenoDB or Ensembl, as it only needs a different path in which to save the results. The whole program has been coded as a shell script, although there is a small part which has been done with Perl, which is the previous step to fastasubseq, in which the start position and the length of the fastasubseq command are saved in the numeros_proteinname file.
The program can be found here. And the Perl program can be found here.