Materiales y métodos
Para conocer las especies con las que compararemos nuestra secuencia nos fijamos en el árbol filogenético de Cebus Capucinus imitator. Este árbol fue generado con Phylot.biobyte disponible online en: (http://phylot.biobyte.de/) [3]
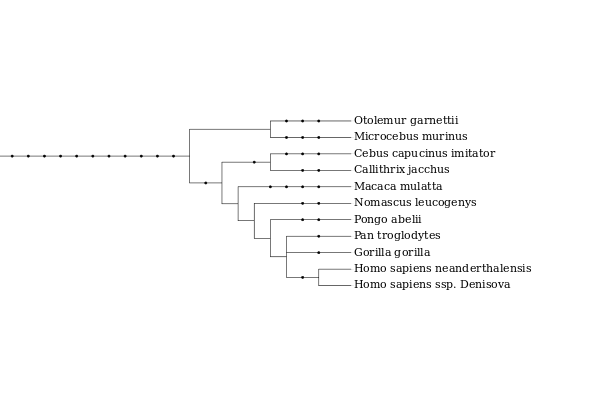
Figure 1. Árbol filogenético que contiene todas las especies SelenoDB y Cebus capucinus imitator.
En la imagen podemos ver las especies más cercanas a Cebus capucinus imitator en SelenoDB. Vemos que la más cercana es Callithrixs Jacchus comúnmente conocido como marmoset en inglés o tití en castellano. Por este motivo, durante nuestro proyecto utilizamos las selenoproteínas de esta especie como querys para encontrar las de nuestra .
Además, durante este trabajo, también hemos querido utilizar las selenoproteínas de Homo sapiens como querys ya que aunque esta sea una especie más lejana a la nuestra está mejor anotada.
Estas proteínas tanto de Callithrixs Jacchur como de Homo sapiens se encuentran divididas en proteínas que contienen la secuencia de selenocisteína, proteínas que contienen los homólogos de cisteína y las proteínas esenciales para la síntesis de selenoproteínas.
Una vez obtenidas las querys se debían cambiar las U (que representaban el codón de selenocisteína) por X manualmente para que el programa pudiera actuar sin problemas.
BLAST (Basic Local Alignment Search Tool) se utiliza para comparar secuencias de aminoácidos o ácidos nucleicos. El programa utiliza un algoritmo heurístico que busca la similitud entre las dos secuencias.
Hay diferentes variedades de BLAST y en este caso utilizaremos T-blastn con el formato 6 para realizar este paso.
T-blastn nos compara secuencias proteicas, lo que nosotros conocemos como querys, con las secuencias nucleotídicas de nuestro genoma de referencia que en este caso es el de Cebus capucinus imitator.
Este comando nos permite obtener hits, es decir secuencias con alineamiento, y seleccionar aquellas con un e-value menor para evitar que este alineamiento se haya producido por azar. En concreto utilizamos un e-value de 10-2.
En el fichero resultante del T-blastn en formato 6 obtendremos la siguiente información esquematizada en la tabla:
| 0 | qsequid | query (e.g., gene) sequence id |
|---|---|---|
| 1 | sseqid | subject (e.g., reference genome) sequence id |
| 2 | pident | percentage of identical matches |
| 3 | lenght | alignment length |
| 4 | mismatch | number of mismatches |
| 5 | gapopen | number of gap openings |
| 6 | qstart | start of alignment in query |
| 7 | qend | end of alignment in query |
| 8 | sstart | start of alignment in subject |
| 9 | send | end of alignment in subject |
| 10 | evalue | expect value |
| 11 | bitscore | bit score |
El comando utilizado para realizar este paso ha sido el siguiente:
"tblastn -query $query_aa.fa -db $genome -out ./$query_aa/BLAST/${query_aa}_blast.fa -evalue 0.01 -outfmt 6"
Fastafetch nos ayuda, una vez sabemos que hits queremos utilizar, a seleccionar los Scaffolds correspondientes. Crearemos un archivo con los identificadores de los Scaffolds con hits de e-value menor al indicado.
El comando utilizado para este paso ha sido el siguiente:
“fastafetch $genome $index '$_' > './$query_aa/fastafetch/${_}.fa' "
Con Fastasubseq, una vez tenemos los Scaffolds, seleccionamos el Scaffold que tiene el e-value más bajo.
Y lo ampliamos para poder trabajar con la selenoproteína completa. Para conseguirlo, se restan 50000 nucleótidos en la posición inicial en caso de que la cadena tenga un sentido positivo, o se suman 50000 nucleótidos si se trata de una cadena con dirección negativa.
Además, en la longitud añadimos 100000 nucleótidos.
El comando utilizado es el siguiente:
Mediante Exonerate y Genewise conseguimos obtener una predicción de los exones que conforman la proteína predicha de Cebus capucinus imitator.
Exonerate nos permite predecir la secuencia que contendría nuestra proteína de interés ya que alinea y compara la secuencia obtenida por Fastasubseq con la secuencia de DNA de la query inicial.
El comando utilizado es:
Nos fijamos que en el comando utilizamos -egrep que nos permite seleccionar únicamente los exones ya que estos son los que finalmente nos darán la proteína de interés. El comando de egrep es el siguiente:
Lo que nos permite hacer Fataseqfromgff es simplemente obtener un archivo con secuencias exónicas en formato FASTA a partir de un archivo GFF. El comando utilizado es:
Fatatranslate utiliza el archivo Fasta anterior que contiene las secuencias exónicas, es decir, el cDNA de las posibles proteínas de interés, para traducirlo a aminoácidos.
Cabe decir que el resultado obtenido contiene todos los aminoácidos posibles a partir de los diferentes marcos de lectura en un archivo multifasta.
El comando utilizado es la siguiente:
T-coffee nos permite hacer un alineamiento global entre las secuencias de aminoácidos obtenidas con fastatranslate y las secuencias de aminoácidos de las querys.
El comando utilizado es:
Con ello se creará un nuevo archivo y si las dos secuencias presentan elevada homología la secuencia predicha será candidata a ser una selenoproteína.
Genewise también hace la comparación de secuencias pero en este caso directamente compara la secuencia de nucleótidos (genoma de referencia) con la de aminoácidos (query).
El comando utilizado es:
Para llevar a cabo el proceso descrito hemos realizado un programa de automatización. (Para obtener el programa clicar aquí)
SECISerch3/Seblastian nos ayudan a confirmar si realmente las predicciones hechas son selenoproteínas o no ya que nos busca los elementos SECIS en la secuencia.
En el caso de selenoproteínas en que hemos hallado elementos SECIS mediante Seblastian, hemos adjuntado los genes que codifican para éstas en la base de datos de SelenoDB en el apartado de Cebus Capucinus (haz click aquí para acceder a SelenoDB).
"fastasubseq './$query_aa/fastafetch/${blast[1]}.fa' $start $length > './$query_aa/fastasubseq/${blast[1]}_fastasubseq.fa' "
"exonerate -m p2g --showtargetgff -q './$query_aa.fa' -t './$query_aa/fastasubseq/$arch' > './$query_aa/exonerate/gff/$arch_prot.gff' "
"egrep -w exon './$query_aa/exonerate/gff/${arch_prot}.gff' > './$query_aa/exonerate/cDNA_gff/${arch_prot}_cDNA.gff' "
"fastaseqfromGFF.pl './$query_aa/fastasubseq/$arch' './$query_aa/exonerate/cDNA_gff/${arch_prot}_cDNA.gff' > './$query_aa/exonerate/cDNApred/${arch_prot}_cDNApred.fa' "
"fastatranslate './$query_aa/exonerate/cDNApred/${arch_prot}_cDNApred.fa' > './$query_aa/exonerate/proteins_gen/${arch_prot}_proteins_gen.mfa' "
"t_coffee $query_aa.fa './$query_aa/exonerate/transcripts/${query_aa}_${marclect}.fa' > './$query_aa/tcoffee/${query_aa}_${marclect}_tcoffee.fa' "
"genewise -cdna -pep -pretty -gff -both './$query_aa.fa' './$query_aa/fastasubseq/$archivo' > './$query_aa/genewise/$protein_file.genewise.fa' "
