METHODOLOGY
The aim of our project was to found out and annotate all the selenoproteins in Nanorana parkeri's genome. Thus, to identify the selenoprotein codifying regions, Xenopus tropicalis was used as a phylogenetic homology-base model. Moreover, in some cases, it was necessary to widen our model, including other genomes of close eukaryotic organisms, mainly Homo sapiens genome which have already been studied for selenoproteins, and it also have been seen that selenoproteins are very conserved. From this point, to initiate our study, Nanorana parkeri's genome that is divided in scaffolds was required to be able to predict all the selenoproteins.
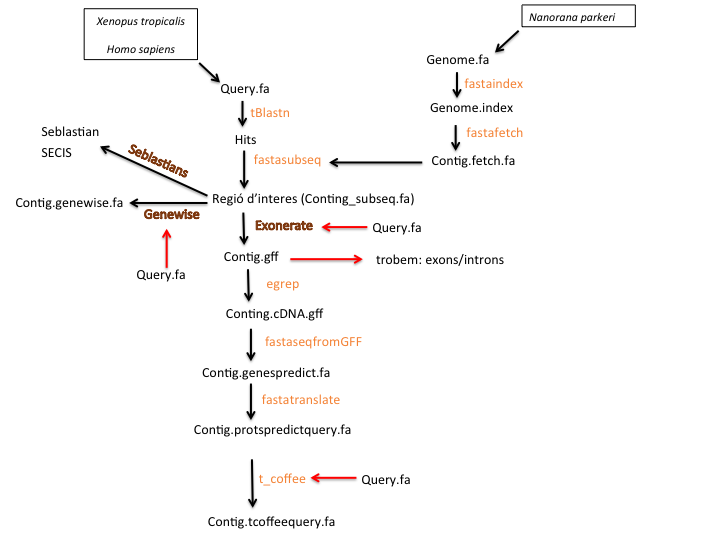
Figure.2.Diagram of the research process of selenoproteins. First of all, we defined our queries, as all the selenoproteins and machinery proteins from Xenopus tropicalis and Homo sapiens. At that moment, we were able to perform the BLAST to find genomic regions with high-homology in our genome, Nanorana parkeri's genome. Before running the gene-structure prediction programs (Exonerate, GeneWise and Seblastian), it is required an index of the genome to speed up all the following procedures and an extraction of the hits subsequences. The cDNA obtained sequences were translated from the exonerate output file, while recruiting the exons. Finally, we achieved a final predicted protein sequence from Nanorana parkeri, which has to be compared with the original query operating the T-coffee. After all, we obtained three analysis outputs: T-Coffe, Genewise and Seblastian. Finally, it was possible to develop a comparison of these results for each query, and also the gene prediction by exonerate was performed for each selenoprotein and machinery protein described.

Queries selection:
As we have already mentioned, the principal reason why we defined Xenopus tropicalis as our reference specie was due to its high phylogenetic relation to Nanorana parkeri and also because proteins, in general, were properly annotated in databases.
It is important to highlight that we achieved a larger part of proteins from SelenoDB, a specific selenoprotein database. Although in most of the cases we were able to find well annotated proteins, in some particular cases, mainly in protein families, we found out that the protein member of the family we were looking for was not correctly defined (its name figured as "NONE"). In those cases, we followed a determined process. First of all, to find the correct name of the protein we performed a blast applying ENSEMBL database. If it did not show proper results, we had to proceed to search for the protein sequence in other known databases, such as UniProt, HomoloGene,UniGene. Finally, if any of these alternative methods were successful, as our last resource, it was necessary to use the human protein to carry out our comparison.
Another minor problem was that sequences from the Selenodb contained the 'U' amino-acids that are not recognized by the exonerate program used afterwards, so we replaced them manually for 'X' in order to avoid problems when using this program.
Genome:
The genome of Nanorana parkeri is available in:
/cursos/BI/genomes/2015/Nanorana_parkeri/genome.fa.
Automation:
Once we obtained the Nanorana parkeri's genome sequence and the selenoprotein queries, to automate the process we created two informatics programs in Perl: programa.sh and programa.pl. The first one, programa.sh, was designed to give the different PATHS to the system to allow the use of several mandatory programmes for the main program to run, and it ends calling programa.pl. The second one, programa.pl, was designed to perform all the commands explained above (except the U to X switch) to predict all the selenoproteins in Nanorana parkeri's genome.
The programs are available in the following link:
PROGRAM.PL
PROGRAM.SH
A part from that, to be able to run the program explained above, all the following files are required to be in the same folder, called master: programa.sh, programa.pl and all the query files, in fasta format, from the different proteins.
To run the program, it is necessary to open the terminal and order the following command:
$ sh ./master/programa.sh
Finally, after running the program, all the achieved outputs will be automatically classified in different folders, which are named as the protein query. Thus, it will be possible to start our protein prediction.
Furthermore, to facilitate the comprehension of our methodology, the main steps of the program will be described in the following sections.
Blast:
To compare each query (selenoprotein from SelenoDB) to our genome we used an algorithm called BLAST (basic alignment search tool), a program for comparing primary biologicas sequence informacion such as amino-acid or nucleotides sequences, enabling to compare a query sequence with a library or database of sequences. We concretely used the command 'tblastn' that aligns the amino-acid sequence of the query with the genome nucleotides of the Nanorana parkeri.
This is the generic command: blastall -p tblastn -i (query) -d (genome) -o query.blast.fa -e 0,05 -m 8
This is our command:
blastall -p tblastn -i $master/$mfile -d /cursos/BI/genomes/2015/Nanorana_parkeri/genome.fa -o ./$outdir/$mfile.blast.fa -e 0.05 -m 8
-p: It specifies the type of search, in this case, tblastn means that we make a protein alignment, so the nucleotide database is translated into aminoacid before the search.
-i: Means input, in this case, it is the query file.
-d: It specifies the database that will be aligned to the input, in this case, the gneome of Nanorana parkeri.
-e: It specifies the maximum e-value allowed, in this case 0,05.
-m: It is used to specify the alignment view; 8: We select which details we eant to see in the output.
The obtained file contains different parameters that allow us to locate the region of the genome that aligns to our queries, such as the length of the sequence aligned, the identity, and the e-value of the alignment, that has to be lower than 0,05.
FastaIndex:
The fastaindex command helps us to optimize the process by indexing the genome of Nanorana parkeri , thus making it much more easy for the program to run and find the searched sequence in the genome.
This is our command:
fastaindex /cursos/BI/genomes/2015/Nanora_parkeri/genome.fa Nanorana_parkeri.index
The input is the non-indexed genome of the Nanorana parkeri, and the output is the indexed genome.
FastaFetch:
This command extracts each scaffold from a given genome. This sequence corresponds to the region that contains the hit, so that we can later extract the exact sequence of the hit by fastasubseq.
This is our command:
fastafetch /cursos/BI/genomes/2015/Nanorana_parkeri/genome.fa Nanorana_parkeri.index '$_' > './$outdir/${_}.fetch.fa'
FastaSubSeq:
This command extracts the hit region that was aligned by the BLAST. The problem is that the alignment only represents the coding part of the genome, that's why we need to extend the search 50000 nucleotides 5' and 50000 nucleotides 3' so as not to miss a part of the sequence because of the introns. If our sequence is not long enough, we will extend the search up to 100000 nucleotides instead of 50000.
fastasubseq './$outdir/${blast[1]}.fetch.fa' $start $length > './$outdir/${blast[1]}.subseq.fa'
Exonerate:
This program will help us to predict the gene and align it with the sequence of the selenoprotein that encodes, and also recognizes the exons, that can be separated from the introns and copied in a single file.
This is the command used:
exonerate -m p2g --showtargetgff -q '$master/$mfile' -t './$outdir/subseq/$file' > './$outdir/exonerate/gff/$protein_file.gff' | egrep -w exon './$outdir/exonerate/gff/$protein_file.gff' > './$outdir/exonerate/cDNAgff/$protein_file.cDNA.gff'
-m: p2g is used to compare the Protein to the Genome.
--showtargetgff: is used to get a file in a GFF format, which contains information about the coordinates.
-q: means that the file is in a FASTA format.
-t: means target, referring to the sequence obtained by the fastasubseq command.
| egrep -w exon: allows us to copy the exonic regions in a file, which format is GFF.
FastaSeqFromGFF:
Using the program FastaseqfromGFF we obtain the cDNA sequence that encodes the final protein. We get it from the subsequence and the file that contains the exons.
This is our command: fastaseqfromGFF.pl './$outdir/subseq/$file' './$outdir/exonerate/cDNAgff/$protein_file.cDNA.gff' > './$outdir/exonerate/genespredict/$protein_file.genespredict.fa'
FastaTranslate:
This program translates the cDNA sequence into the final protein.
This is our command: fastatranslate -f './$outdir/exonerate/genespredict/$protein_file.genespredict.fa' -F 1 > './$outdir/exonerate/protspredict/$protein_file.protspredict.fa'
-f: is used to specify the file containing the exons in a nucleotidic format.
-F: is used to specify the reading frame (3 forward and 3 reverse), in this case -F1.
T_coffee:
This command is used to compare two sequences, in this case we compare the known sequence (query protein) with the homologous sequence of the Nanorana parkeri (predicted protein).
This is our command:
t_coffee './$master/$mfile' './$outdir/exonerate/protsdef/$protein_file.protsdef.fa' > './$outdir/tcoffee/$protein_file.tcoffee.fa'
Genewise:
This program is also for gene prediction for homologous sequences, so we used it to contrast the data obtained by the exonerate program.
This is our command:
genewise -cdna -pep -pretty -gff -both './master/$mfile' './$outdir/subseq/$file' > './$outdir/genewise/$protein_file.genewise.fa'
-cdna: is used to show the predicted cDNA sequence aligned.
-pep: is used to show the predictet peptid.
-pretty: is used to show the ASCII alignment.
-gff: is used to create the output in a gff format.
-both: is used to scan in a forward and reverse way as the genewise scans forward automaticaly.
Protein assignation:
After running our program, it was required to analyze all the obtained results. First of all, we needed to relate the hits achieved for a concrete protein. In this first step, we take into account some specific features of each hit to select the best possible one. In the blast file we tried to find the best hit considering the lowest e-value, a sufficient homology and an appropriate length. Moreover, in case of protein families, we found the same scaffolds associated to distinct proteins in the same family due to the intrafamiliar homolgy. In these special cases, we had to analyze each protein member of the family through choosing the best hit following the general steps explained above, eliminating this scaffold of the list of possible scaffolds for the other proteins of the family, which often has worst e-values for the other proteins of the same family. In addition, in family cases, a phylogenetic tree of the family's proteins was performed to clearly define the best hit. Finally, it was possible to determine the protein-containing scaffold.
At that point, we were able to analyze the gene location and also the number of exons and introns from the exonerate files of each protein. Hence, from our predicted protein file it was possible to analyze the Sec- or Cys-containing residue.
At the end, analyzing the t_coffee file we were able to make a comparison between the predicted protein, from the elected hit, and the initial protein from Xenopus tropicalis or Homo sapiens. Thus, this alignment reflects the proper prediction of the protein, and also in those cases in which our predicted protein was found in different scaffolds.
SECIS and Seblastian:
SECIS search 3 is an in silico method that allows us to identify SECIS elements in the eukaryotic organisms. This is an extremely sensible method, but its specificity is not really high because it is only based in the input sequence regardless the fact that the SECIS element has to be in the 3'UTR region; that is why a lot of false positives were found. Thus, when it was possible, we proceed to use the Seblastian program to determine them. Seblastian is an in silico method that detects selenoprotein genes that are found upstream a SECIS element, so it uses SECIS search 3 itself. It was very useful to confirm or refute the SECIS elements previously found, and we only accepted those that were next to a selenoprotein gene found by the Seblastian program.