
L'any 2001 es va publicar la seqüenciació del genoma humà. A partir d'aleshores, es van començar a analitzar les seves regions amb les eines bioinformàtiques del qual es disposaven; per poder-ne desxifrar els seu significat. Amb aquesta finalitat, es va crear el projecte ENCODE ( ENCyclopedia Of DNA Elements ) i així identificar els elements funcionals de les seqüències del genoma humà. La fase pilot d'aquest projecte sols ocupa un 1% del genoma humà ( unes 30 Megabases ).
El nostre treball es basa en l'anàlisi d'una de les seqüències genòmiques que formen part d'aquest projecte ENCODE. Per tant, l'objectiu principal és intentar caracteritzar una regió anònima, anomenada ENr123; mitjançant les eines de la bioinformàtica, dins les limitacions dels nostres ordinadors i dels nostres coneixements.
![]()
El primer pas, i el més important, és obtenir la seqüència que volem analitzar. Per tant, anem a la pàgina web del projecte ENCODE i ens la descarragem.
La nostra seqüència és la ENr123, es troba en el braç q del
cromosoma 12 i té una mida de 500.000 bases.
A continuació teniu una repesentació d'aquesta regió :

La seqüència es guarda en format fasta : ENr123.fa
Un cop ja tenim la seqüència en un fitxer, haurem de comprovar si conté els nucleòtids que estàven anotats. Per fer això, es modifica el format fasta i es tabula el fitxer per facilitar el recompte. La transformació de l'estructura es realitza amb les comandes bàsiques del Unix:
awk '{printf $1}' ENr123.fa > ENr123.tbl
També es compta el número de nucleòtids de la seqüècia amb la següent
comanda :
awk '{print length($2)}' ENr123.tbl .
El resultat és de 500.000
bases, justament el que esperàvem.
Abans, de dur a terme cap tipus de predicció de gens ni d'estudiar els
possibles elements traduccionals que hi pugui haver dins de la nostra regió,
s'ha de calcular el percentatge de G i C que té la seqüència. Aquesta informació
és molt útil perquè a les illes G-C és on més probabilitat hi ha de trobar gens.
És per això que aquesta informació ens donarà una idea dels resultats que obtindrem al
llarg del treball.
La comanda que es fa servir en aquest cas és :
awk '{print $2}' ENr123.tbl
| fold -1 | sort | uniq -c | gawk '{print $2,$1/500000}'
I el resultat que obtenim és:
A l'observar aquests resultats, podem predir que menys de la meitat de la
seqüència que estem analitzant serà codificant. Sols esperem que codifique per a
gens un 36% dels 500.000 nucleòtids.
Un cop analitzat a grans trets la nostra regió del genoma, passarem a
analitzar el contingut genòmic que puga haver-hi.
![]()
A continuació,després de tindre la seqüència en format Fasta i haver comprovat el nombre de bases que conté, passarem a emmascarar-la.L'objectiu de l'emmascarament és poder eliminar tots aquells elements repetitius que hi ha a la seqüència per evitar que interferisca en els resultats de la predicció de gens. Per poder realitzar l'emmascarament, hem d'anar al servidor que RepeatMasker ens ofereix.
Les condicions que apliquem per a dur a terme l'emmascarament són:
El resultat són aquests 4 fitxers que hi ha a continuació, cadascun dels quals ens representa una informació diferent:
A partir d'ara, tot el treball el realitzarem a partir de la seqüència emmascarada.
Un cop ja tenim tots els arxius, farem una represenatció per poder
visualitzar quins sn els dominis repetitius més abundants i quines són les
seves localitzacions.
Per poder transformar el fitxer de text en una imatge, hem de convertir el
fitxer que est` en format "seq.out" en un fitxer en format "seq.gff". La comanda
que utilitzarem en aquest cas és un xicotet programa en 'gawk':
export PATH=/disc8/soft/perl/bin/:/disc8/bin/:$PATH
grep ENr123 ENr123slow.seq.out | \
awk 'BEGIN{ OFS="\t" }
{ print $5, $11, "repeat", $6, $7, ".", ".", "."; }
' > ENr123slow.seq.out.gff
Ara sols cal utilitzar els programes de visualització per poder obtindre la
representació que esperem :
Aquesta és la imatge de les repeticions i les seves distribucions:
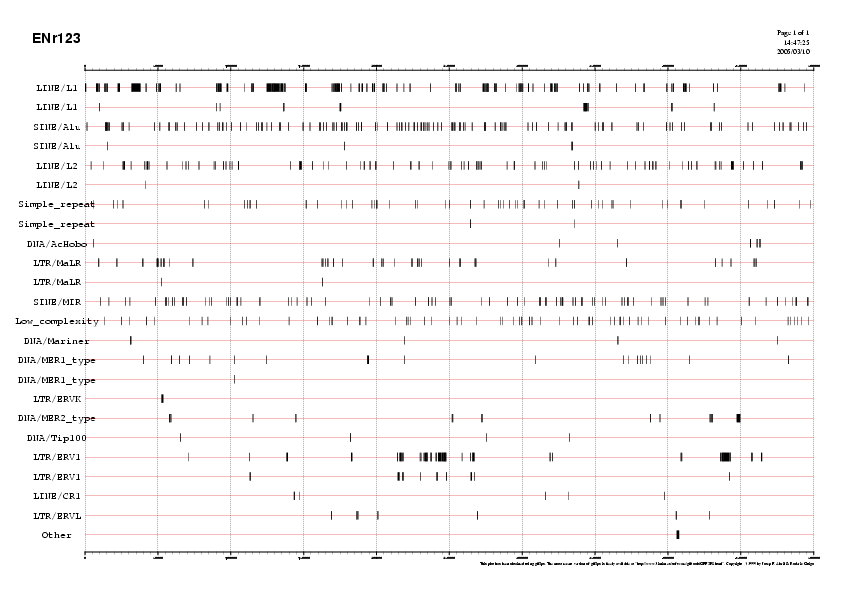
En aquesta representació podem observar que la nostra seqüència conté grans regions repetitives LINE i SINE. A més, les repeticions simples i les de baixa complexitat es troben homogèniament distribuides per la llargària de la seqüència. La resta d'informació s'analitzarà en l'apartat de conclusions.
Ara ja tenim emmascarada la nostra regió, Seqüència.masked , aleshores els següents pasos que es realitzen al treball, es faran prenent-la com a seqüència de partida.
![]()
Ha arribat el moment de dur a terme les prediccions dels possibles gens que hi ha en la nostra regió, fent servir 3 programes de prediccions de gens. Cadascun dels programes utilitzen una matriu de pesos diferent per predir les regions codificants. és per això que es prediuen els gens en tres programes diferents, per poder realitzar una comparació més acurada. Els tres programes utilitzats són : GENEID, GENSCAN i FGENESH .
La primera predicció que duem a terme és la del Geneid. Les condicions que s'utilitzen estàn especificades a continuació:
- No representació d'imatge - Homo sapiens - Prediction mode normal - DNA strands forwar&reverse - Sense senyals ni exons marcats
El resultat s'obté en format gff : Prediccio_geneid.gff
Per poder visualitzar la imatge d'aquesta representació seguim els següents passos:
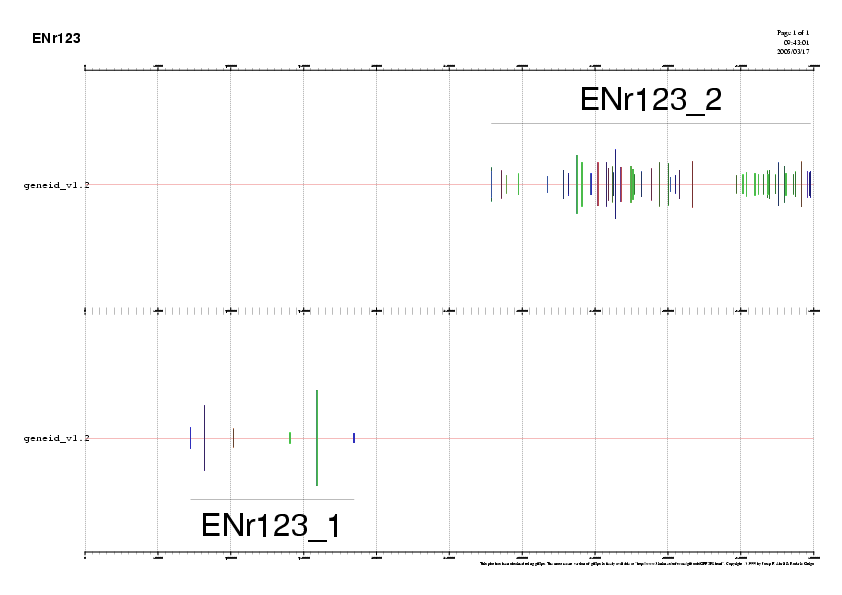
La informació de la predicció està en aquesta taula:
| Nm. de gen | Nm. d'exons | Posicio Inicial | Posicio Final | Direccio | Nm.aminoacids |
| 1 | 6 | 184.634 | 72.439 | - | 337 aa |
| 2 | 51 | 278.725 | 497.909 | + | 2.656 aa |
En la següent predicció utilitzarem el programa GENSCAN .
Aquest servidor té un límit de bases que pot predir, per això que hem de fer petites modificacions en la seqüència query. Hem de tallar-la en segments de 100.000 bases i així predir els gens a partir diferents trossos de la seqüència. Un cop tinguem la predicció, haurem de reconstruir la informació per tal de poder interpretar el nombre total de gens que hi ha en els 500.000 nucleòtids. Anirem, doncs, per parts.
Aquesta es talla en fragments de 100.000 bases amb un
solapament de 10.000. La presència d'aquesta superposició ens facilita la
posterior manipulació del resultat. Com que en cada regió hi ha 10.000 bases
que coincideixen, suposem que en aquestes regions ens haurà predit els
mateixos exons. Un cop ja tinguem la predicció haurem de triar quina seria la
reconstrucció dels gens més fiables.
Per poder la tallar la seqüència utilitzem la funció del software
Fastachunk. Amb aquesta comanda vam generar 5 fitxers nous:
Primerament cridarem la funció del software:
export
PATH=$PATH:/disc8/bin
I a continuació obtindrem les 5 regions:
Després d'haver tallat les diferents regions, es modificarà la seva estructura transformant-la en seqüències FASTA ( es modifica amb l'emacs posant a la primera línia el símbol " >Enr123 " ).
Les opcions que vam utilitzar en aquest programa són:
- Vertebrats - Score = 1.00 - Predicted CDS and peptidsEl resultat de l'anàlisi es van enregistrar en els segents fitxers:
Per poder treballar amb aquests fitxers és necessari homegeneitzar les dades i convertir l'output que el servidor genscan ens ha ofert en un fitxer gff. Les comandes que es fan servir en cadascun dels subfitxers són :
gawk 'BEGIN{OFS="\t"}
$2 ~ /Term|Intr|Init/ {
print "ENr123_1", "genscan", $2, start=($4<$5 ? $4 : $5),
end=($5<$4 ? $4 : $5), $13, $3, $7,"Seq1_"$1"";
}' genscan100000.txt | \
sed 's/\.[0-9][0-9]$//' > ENr123_100000.genscan.gff
gawk 'BEGIN{OFS="\t"}
$2 ~ /Term|Intr|Init/ {
print "ENr123_2", "genscan", $2, start=($4<$5 ? $4 : $5),
end=($5<$4 ? $4 : $5), $13, $3, $7, "Seq2_"$1"";
}' genscan200000.txt | \
sed 's/\.[0-9][0-9]$//' > ENr123_200000.genscan.gff
gawk 'BEGIN{OFS="\t"}
$2 ~ /Term|Intr|Init/ {
print "ENr123_3", "genscan", $2, start=($4<$5 ? $4 : $5),
end=($5<$4 ? $4 : $5), $13, $3, $7, "Seq3_"$1"";
}' genscan300000.txt | \
sed 's/\.[0-9][0-9]$//' > ENr123_300000.genscan.gff
gawk 'BEGIN{OFS="\t"}
$2 ~ /Term|Intr|Init/ {
print "ENr123_4", "genscan", $2, start=($4<$5 ? $4 : $5),
end=($5<$4 ? $4 : $5), $13, $3, $7, "Seq4_"$1"";
}' genscan400000.txt | \
sed 's/\.[0-9][0-9]$//' > ENr123_400000.genscan.gff
gawk 'BEGIN{OFS="\t"}
$2 ~ /Term|Intr|Init/ {
print "ENr123_5", "genscan", $2, start=($4<$5 ? $4 : $5),
end=($5<$4 ? $4 : $5), $13, $3, $7, "Seq5_"$1"";
}' genscan500000.txt | \
sed 's/\.[0-9][0-9]$//' > ENr123_500000.genscan.gff
Per poder concatenar tots els segments i així poder predir de manera uniforme tots els exons que hi ha a la nostra regió ENr123, hem de posar coordenades absolutes als principis i finals d'exons, prenent com a referència la base 0. És a dir, hem de sumar el nombre de bases que abans li hem restat per tallar les diferents subseqüències ( en el primer segment de 0 a 100000 bases no cal realitzar el càlcul ).
En aquest cas, utilitzarem un programa en 'gawk' que ens permet fer la transformació immediata:
Tros de 90000 fins 200000:
gawk 'BEGIN{OFS="\t"} {$4=$4+90000 - 1;$5=$5+90000 - 1;print}' ENr123_200000.genscan.gff > ENr123_200000.abs.genscan.gff
Tros de 190000 fins 300000:
gawk 'BEGIN{OFS="\t"} {$4=$4+190000 -1;$5=$5+190000 - 1;print}' ENr123_300000.genscan.gff > ENr123_300000.abs.genscan.gff
Tros de 290000 fins a 400000:
gawk 'BEGIN{OFS="\t"} {$4=$4+290000 - 1;$5=$5+290000-1;print}' ENr123_400000.genscan.gff > ENr123_400000.abs.genscan.gff
Tros de 390000 fins a 500000:
gawk 'BEGIN{OFS="\t"} {$4=$4+390000 - 1;$5=$5+390000 -1;print}' ENr123_500000.genscan.gff > ENr123_500000.abs.genscan.gff
Després d'haver fet algunes proves amb aquests resultats, ens vam adonar que els frames calculats per aquest programa no coincidien amb els que ens predia GENEID. Per això hem de transformar els frames per facilitar-nos la feina de recomposició dels exons.
Vam utilitzar un xicotet programa en 'awk':
#!/bin/gawk -f
### Per corregir les frames del genscan...
$1 !~ /^\#/ {
if ($7 == "+") {
if ($3 == "Init") {
L = 0;
};
l = $5 - $4 + 1;
F = (3 - (L % 3)) % 3;
print $1,$2,$3,$4,$5,$6,$7,F,$9;
L = L + l;
}
else {
if ($3 == "Term") {
L = 0;
};
l = $5 - $4 + 1;
L = L + l;
F = L % 3;
print $1,$2,$3,$4,$5,$6,$7,F,$9;
};
}
per canviar els frames a partir dels fitxers en els quals ja tenim les
coordenades en valor absolut.
El resultat en "gff" es pot visualitzar:
Regió 0 a 100000 bases: genscan100000_frames.gff Regió 90000 a 200000 bases: genscan200000_frames.gff Regió 190000 a 300000 bases: genscan300000_frames.gff Regió 290000 a 400000 bases: genscan400000_frames.gff Regió 390000 a 500000 bases: genscan500000_frames.gff
En aquest moment, tenim les prediccions dels exons de les 5 regions que hem tallat inicialment, amb totes les coordenades canviades respecte a la base 0 i amb el càlcul del frame correctament. Ara toca cancatenar totes les regions i poder reconstruir la predicció de tots els exons i gens que el programa ens ha predit.
Per poder concatenar tots els fitxers utilitzem aquesta comanda i generem un nou fitxer:
cat genscan.abs_frames.gff genscan200000_frames.gff
genscan300000_frames.gff genscan400000_frames.gff genscan500000_frames.gff |
sort +3n > genscan.complet_frames.gff
Aleshores, en aquest fitxer tenim totes les prediccions i la reconstrucció
que finalment hem realitzat.
Quan analitzem els exons predits entre les
regions solapants, observem que hi ha algunes prediccions que no coincideixen.
Açò ens obliga a adoptar uns criteris per triar els exons que formaran part de
la reconstrucció final.
Els solaplaments es trobaven entre les segents regions :
## ENr123_4 genscan: Init 290306-290351 de Seq3_2 # Seq4_1 :
s'elimina perquè no coincideix amb la pauta de lectura de l'exó anterior ni
amb el posterior.Probablement, no siga una bona predicció ja que és el
primer exó predit en la 4a regió i com que ha de començar un exó, agafa el
primer que troba.
## ENr123_3 genscan: Intr 291561-291695 de Seq3_2 : s'elimina
perquè igual que a l'exó de la predicció feta en la regió 4 i, a més a més,
la seva eliminació no trenca la pauta de lectura.
## ENr123_3 genscan: Intr 297142-297239 de Seq3_2 : s'elimina
perquè tot i no coincidir exactament amb el final d'exó predit en la regió
4, la diferència de bases no fa modificar la pauta de lectura. Escollim
aquest exó degut a què a partir d'ací ja utilitzem tots els exons predits en
la regió 4.
Ara, doncs, després d'haver eliminat aquest 4 exons corresponents al 3r gen predit per aquest programa, comprovarem que l'eliminació no afecta a la pauta de lectura dels exons.
La comprovació de la conservació de la pauta de lectura la realitzem traduint els exons problemàtics als aminoàcids que codifiquen cadascun d'ells. El resultat, per considerar-lo correcte, no haurà de tenir cap codó STOP al mig de la seqüència.
Primer, hem d'extraure del fitxer ENr123.fa, els fragments corresponents als exons eliminats en la regió de solapament 290000 a 300000. En una taula, escriurem quins fragments sn els que haurem de tallar: exons1_7.tbl
Posteriorment, utilitzarem la comanda :
Així ens crea 7 fitxers diferents on tindrem els nucleòtids corresponents a aquests exons : genscan.Exo1.fa,genscan.Exo2.fa,genscan.Exo3.fa,genscan.Exo4.fa,genscan.Exo5.fa,genscan.Exo6.fa, i genscan.Exo7.fa .
Ara, per poder dur a terme la traducció als aminoàcids, haurem de concatenar els 7 fitxers:
cat genscan.exons1_7.Exo_1.fa genscan.exons1_7.Exo_2.fa
genscan.exons1_7.Exo_3.fa genscan.exons1_7.Exo_4.fa genscan.exons1_7.Exo_5.fa
genscan.exons1_7.Exo_6.fa genscan.exons1_7.Exo_7.fa | perl -e
'while(
Per observar la traducció als aminoàcids gastarem els programa en 'perl' genscan.aa.pl. I el resultat de l'execució del programa es guarda en aa.exonsjunt.txt. En aquest fitxer podem observar com no apareix cap cod STOP al traduir aquesta porció del gen.
Desprè:s de totes aquestes comprovacions, podem concluir que la nostra recomposició dels gens no afecta a la pauta de lectura dels mateixos.
La visualització consisteix en la tranforamció del fitxer "gff" en un de "png" per poder visualitzar-ho amb el programa kview :
La imatge que podem observar és la següent:
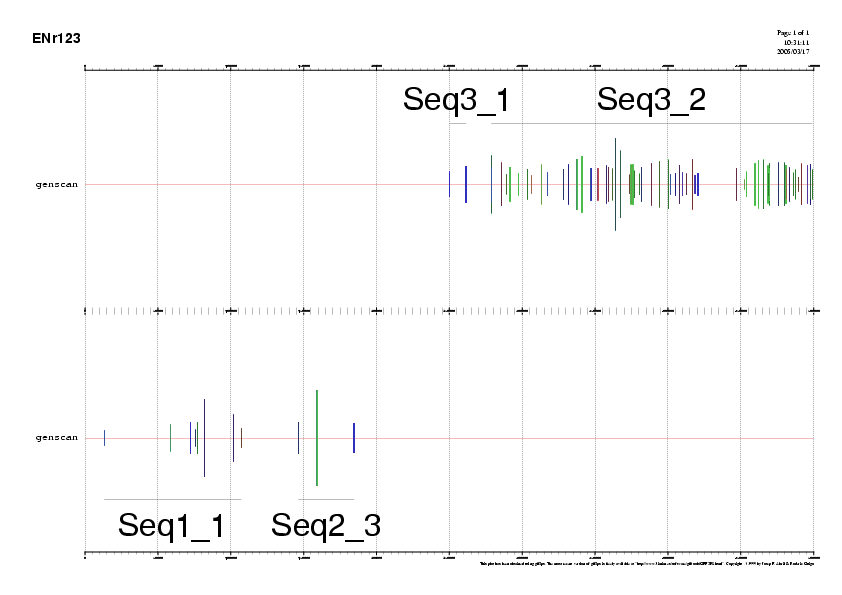
Observant a gran augment aquesta imatge, podem comprovar com el frame de cada exó és correcte ( es mira si conincidixen els colors entre el 3' i 5' de cada exó). Aquesta és una altra indicació que la reconstrucci&oacte; del gen l'hem fet correctament.
A l'igual com hem fet en la pedicció amb el programa GENEID, i per donar per finalitzada aquesta predicció, podem esquematitzar els resultats obtinguts en la següent taula :
| Nm. de gen | Nm. d'exons | Posicio Inicial | Posicio Final | Direccio |
| 1 | 9 | 107.232 | 13.253 | - |
| 2 | 3 | 184.633 | 146.413 | - |
| 3 | 2 | 250.261 | 261.553 | + |
| 4 | 69 | 278.724 | 499.236 | + |
Per últim, utilitzarem el servidor de FGenesh per obtenir una tercera
predicció de gens.En aquest cas, el servidor ens admet la seqüència sencera de
500.000 bases i no caldrà fer tants passos com hem fet en el pas anterior.
El resultat que n'obtenim és : FGenesh.txt
Per aquesta predicció no ens ha donat el frame de cada ex. És per això que hem fet un petit programa en 'awk' per aconseguir-lo : FGenesh.awk . Cal esmentar que podiem haver obviat aquesta dada ja que no és imprescindible per a la transformació de l'output en "gff", per nosaltres l'hem calculat per tindre una millor predicció.
Per executar aquest programa es fa servir la segent comanda :
./fgnenesh2gff.awk < FGenesh.txt > FGenesh_withframes.gff
I per poder visualitzar la imatge, hem fet el següent :
La visualització de la predicció d'exons és aquesta:
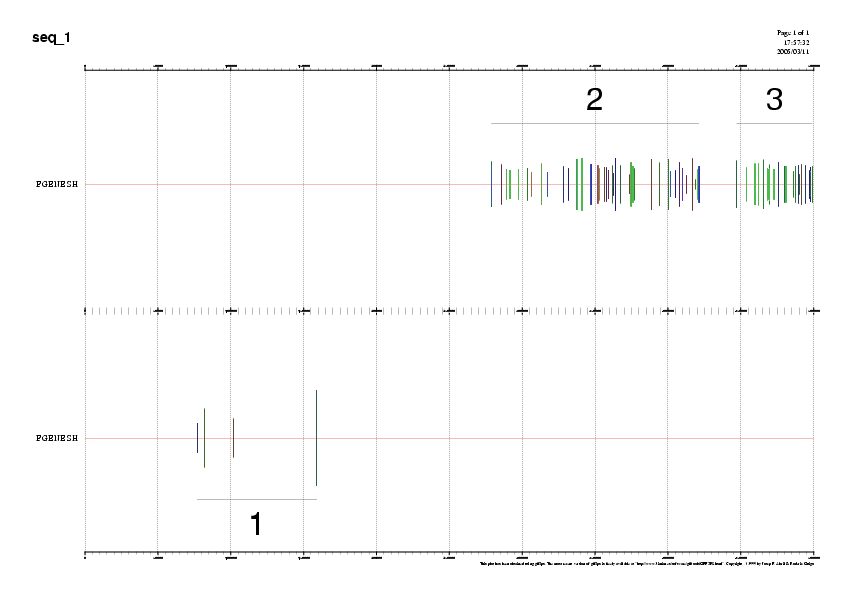
I, com ja hem fet en les tres anteriors representacions, esquematitzem en una taula el resultat de la predicci:
| Nm. de gen | Nm. d'exons | Posicio Inicial | Posicio Final | Llargada | Direccio | Nm. aminoacids |
| 1 | 4 | 159.086 | 76.914 | 756 pb | - | 251 aa |
| 2 | 43 | 278.725 | 421.358 | 6.336 pb | + | 2111 aa |
| 3 | 25 | 447.041 | 499.237 | 3.423 pb | + | 1144 aa |
Finalment, podem comparar les tres prediccions obtingudes amb el Geneid, el
Genscan i el FGenesh. A partir dels 3 documents en format "png" obtinguts
anteriorment, representem en una única imatge formada per les 3 prediccions. Les
comandes utilitzades en aquest cas són :
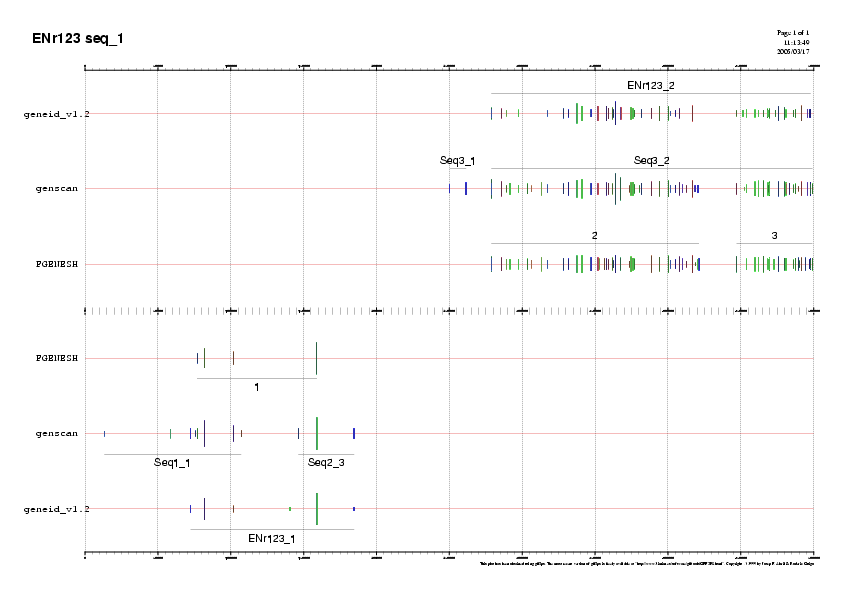
Es pot observar, que en cada programa ens ix una predicció diferent, cosa que
ens obliga a fer una validació de les prediccions. D'aquesta manera podrem
escollir la predicció més acurada per esbrinar quina és la proteïna
que codifica.
![]()
Per poder assolir aquest objectiu, compararem la seqüència query contra la base de dades d'EST humans Megablast, programa que ens ofereix els servidor NCBI BLAST.
Abans de realitzar la comparació, per poder tindre una millor resolució,
tallem la nostra seqüència en dues porcions que engloben els 3 gens predits. La
divisió està basada en la clara separació de gens reverse vs forward.
Així, tenim una primera regió que inclou els gens predits en reverse
( un gen predit per FGENESH i GENEID i dos predits per GENSCAN ).I una segona regió pels gens
predits en forward ( un segons GENEID i dos segons FGENESH i GENSCAN ).
L'estructura que hem elegit per tallar les regions es pot observar en la
següent imatge:
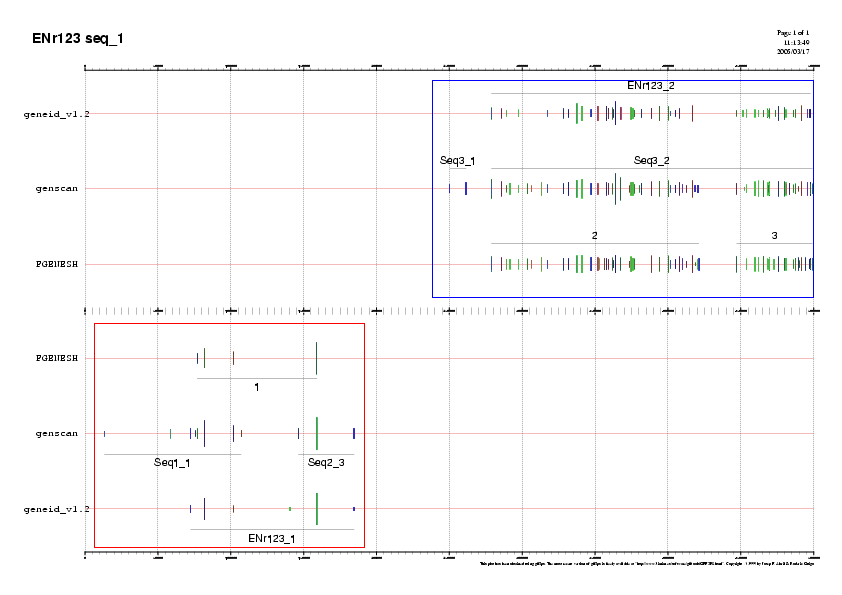
Per començar, busquem les coordenades de totes les prediccions per poder incloure des de la base més xicoteta fins a la més gran. A continuació, s'especifica quin ha estat el límit de predicció en els diferents programes i com s'han tallat les dues porcions.
Per deixar un marge d'error, tallem la seqüència emmascarada des de la base 12000 fins a la 190000 :geneid: 72430 - 184634 genscan: 13253 - 184634 FGenesh: 76914 - 1590086
export PATH=$PATH:/disc8/bin
fastachunk ENr123slow.seq.masked 12000
178000 | fold -60 > gen1.fa
Per deixar un marge d'error, tallarem la seqüència emmascarada des de la base 278000 fins 500000:geneid: 279148 - 497909 genscan: 250333 - 499237 FGenesh: 278725 - 499237
export PATH=$PATH:/disc8/bin
fastachunk ENr123slow.seq.masked 278000 222000 | fold -60 > gen2.fa
Ara, passem a comparar-les amb la base de dades EST que abans ja haviem introduit.
- EST_HUMAN - Low complexity, Human repeats, Mask for lookup table only - Word size : 11 - % Identity : 98,1,3 - Pairwise - Format at the bottom - Alignements : 50
Els resultats d'aquesta validació es poden visualitzar a
regio1.EST.html i regio1.EST.txt.
Per analitzar millor el resultat obtingut en "txt", el transformarem a un fitxer "gff" i així
visualitzar-lo. Emprem :
export PATH=/disc8/soft/perl/bin/:/disc8/bin/:$PATH
parseblast.pl -Gi gen1.prova.blast.est.txt > gen1.prova.blast.est.gff
Però, no ens oblidem que hem de transformar les coordenades de la regió
respecte a l'absoluta, per poder comparar la distribució dels EST respecte als exons
predits:
gawk 'BEGIN{OFS="\t"} {$4=$4+ 12000 - 1;$5=$5+12000 - 1;print}' gen1.prova.blast.est.gff > regio1_abs.EST.gff
Un cop transformades les coordenades, filtrarem els EST obtinguts per poder eliminar aquells que no tinguin una alta coincidència. Per poder dur a terme la tria farem gastar el programa:
BEGIN{
OFS="\t";
}
{
nhsp[$9]++;
hsp[$9,nhsp[$9]]=$0;
}
END{
for (i in nhsp)
if (nhsp[i]>1)
for (j=1;j<=nhsp[i];j++)
print hsp[i,j];
}' gen1_abs.prova.blast.est.gff > regio1.tria.EST.gff
Per verificar que el nombre de EST ha disminuit després d'haver fet la tria, hem de comptar el contigut de EST en cadascun dels fitxers i així verifiquem que de 72 EST predits n'hem pogut filtrar un 50% i quedar-nos nom&eacte; en 35 EST. Un cop ja tenim totes les dades, podrem visualitzar la predicció dels exons juntament la validació dels mateixos:
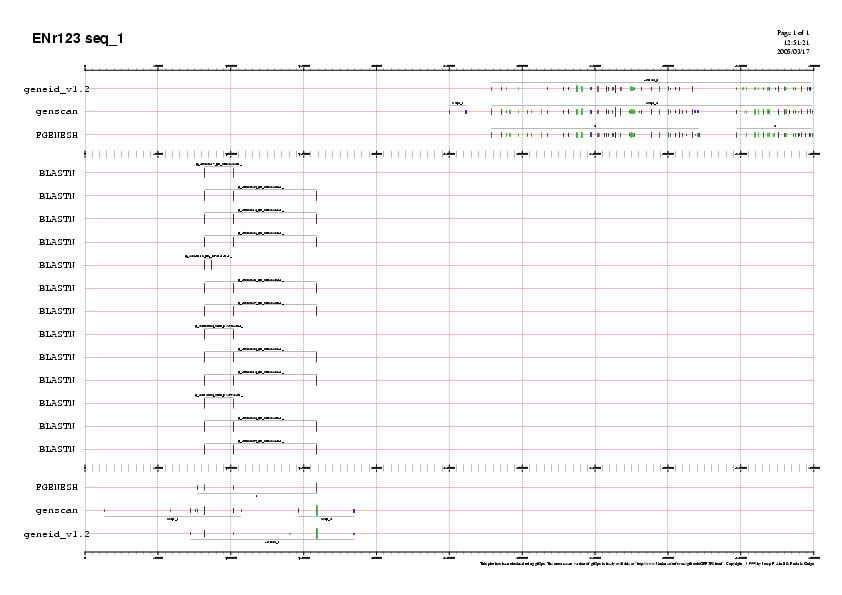
- EST_HUMAN - Low complexity, Human repeats, Mask for lookup table only - Word size : 16 - % Identity : 99,1,3 - Pairwise - Format at the bottom - Alignements : 50
I els resultats els guardem en els fitxers gen2.EST.html i
gen2.EST.txt.
Igual que abans, amb el programa Parseblast, el fitxer "txt" es transforma a un fitxer "gff"
i el modifiquem per poder tindre les coordenades absolutes. Com que els passos
són exactament els mateixos que els anteriors, directament adjuntem el fitxer "gff"
amb les coordenades absolutes i després filtrem,
regio2.tria.EST.gff.
Altra vegada, el resultat es redueix dràsticament, de 77 EST predits, passem a
tindre 43 EST seleccionats.
Ara, per poder visualitzar la imatge dels EST juntament amb la predicció dels gens.
Els EST ens queden al mig de la imatge. Hem de modificar el fitxer "gff" llevant-li
la orientació dels EST:
gawk '{$7="."; print $0}' tria.est.gen2.gff > regio2.tria_noframes.gff
Aquí, es representa les prediccions dels gens amb la validació amb els EST:
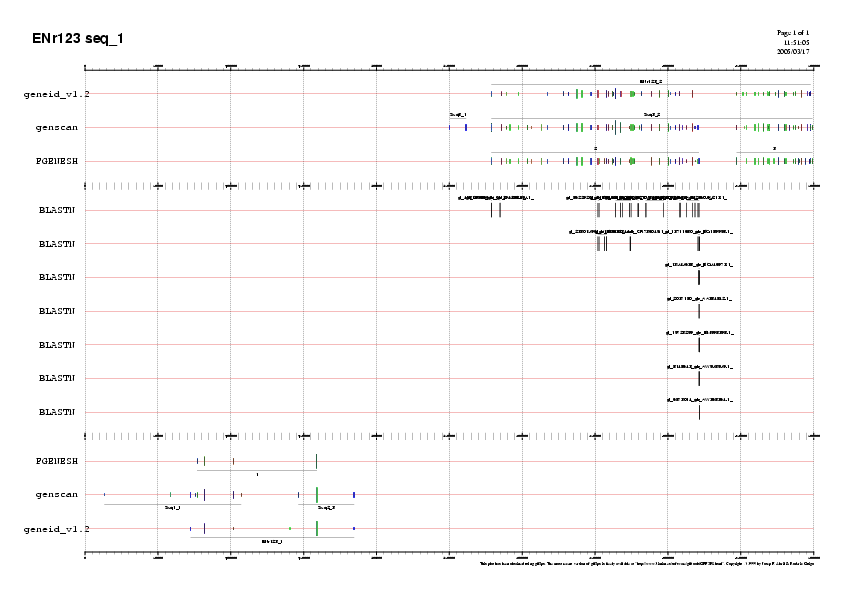
Ara podem visualitzar les prediccions EST, tant de la regió 1 com la 2, juntament amb totes les prediccions dels gens, i així tenim una visió més global:
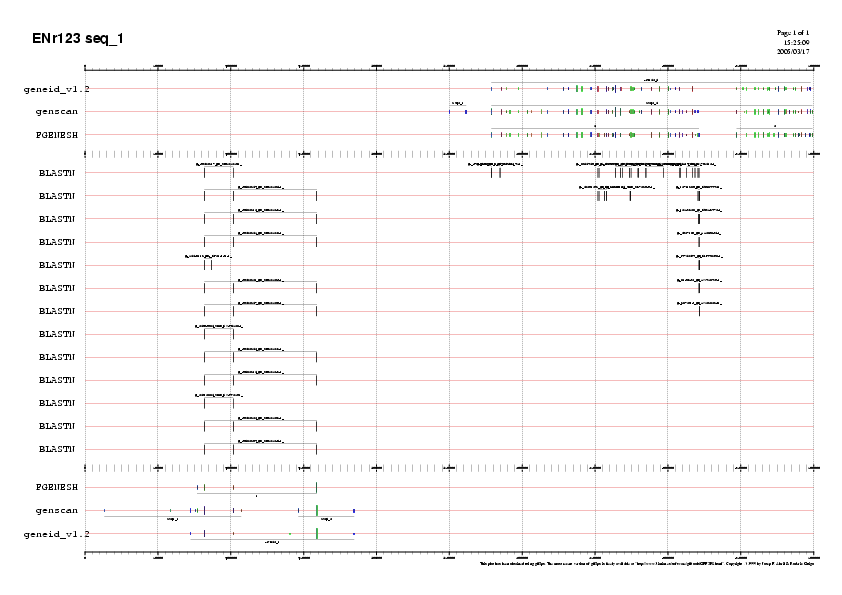
Fins a aquest moment, hem analitzat els EST que suportaràn alguna de les prediccions
dels exon realitzats anteriorment. Serà l'hora de passar a avaluar quins són
els gens que ens creguem com a certs, segons la similitut amb la base de dades d'EST.
![]()
Gràcies a la comparació de la nostra seqüència amb les bases de dades de EST humans,
hem pogut validar les prediccions que haviem fet. Hem assumit que la nostra regió té
4 gens.
Observant la imatge on tenim les tres prediccions de gens i validació per EST,
podem veure que els EST ens estàn suportant la presència de 3 gens de manera clara.
Els EST són procions de cDNA que codifiquen per a regions codificants d'exons ja
estudiats al genoma humà. La síntesi d'EST es realitza a partir de la regió
poliA, és per això que la majoria d'EST es localitzen a la regió 3' dels
gens.
Un cop explicat la funció dels EST, podem afirmar que els EST ens recolzen els
següents gens:
Un cop ja analitzades les validacions i haver triat els gens, anirem als documents "txt" de cadascuna de les prediccions que hem escollit, i capturarem la seva seqüència aminoacídica.
En aquest cas, hem trobat un bon aliniament, del 92% d'identitat i amb un SCORE molt baix : e-109. La seva funció és un transportador de myo-inositol.

Utilitzant la mateixa base de dades, busquem els diferents dominis que es puguin alinear
correctament amb aquestes regió. Proteina_2
Els resultats obtinguts en aquest BLAST ens verifiquen que aquesta predicció
no és real. La màxima similaritat que prediu aquesta base de dades és
del 45%, per això no podem predir cap funció, no seria significativa.
Els resultats es poden observar a gen2.blast.html i
gen2.blast.txt.
Així podem afirmar que realment la predicció d'EST ens estava indicant
que açò no era un gen.
Amb la proteïna corresponent al gen 3, proteina_3.txt,
farem un BLASTp, igual com hem realitzat amb els anteriors gens.
En aquest cas realitzem 2 blasts, un corresponent a la base de dades swissprot
i l'altra amb la condició de nr.
Aquestes dues prediccions les hem realitzat perquè els resultats del swissprot,
molt més rígides que els de nr, ens impedia vore alguna
proteïna que tinguera una similaritat major al 50%.
Els resultats obtinguts en la condició nr es poden visualitzar
a prot3.blast.html i prot3.blast.txt.
A partir d'aquestes dades, podem predir que aquest gen correspon a una leucine-rich repeat
kinase 2. La identitat que representa aquest gen amb aquest proteïna de la base de
dades és d'un 78% d'identitat.
La representació gràfica d'aquest domini no l'hem pogut trobar per enlloc
de la base de dades.
D'aquest gen en tenim l'inici però no podem saber on acaba perqué
està dins de la següent regió del projecte ENCODE. Així el que
farem serà intentar predir un domini funcional que puga encaixar
amb aquesta porció del gen.
Aquesta regió, proteina_4.txt, s'extrau de la
predicció FGenesh.
Realitzem el BLAST i obtenim els resultats, tant en el paràmetre nr com en el swissprot,
perquè, com en el cas anterior, necessitem tindre un alinemament que siga
estadísticament significatiu. Basant-se en els resultats de les condicons nr,
prot4.blast.html i prot4.blast.txt, podem afirmar que aquesta regió correspon a un domini similar to submaxillary apomucine amb identificador del GeneID=441636.
Tot i haver trobat la homologia i observar que els e-scores són molt xicotets,
és a dir, deuen de ser bones presiccions, no trobem cap
respresentació del domini.
Aquest fet ens obliga a anar a la base de dades Interpro
per poder verificar que aquest domini predit existix. Els resultats obtinguts
d'aquesta predicció la guardem en
prot4_interpro.html.
Hem buscat en una nova base de dades, per&oagrave; els resultats ens indiquen que
corresponen al mateix domini abans descrit. Els scores tan elevats serien ocasionats
a l'alta repetició que existix en aquesta regió. Aquests dominis conserven un
alt grau de repeticions. Així acceptem que el nostre gen és una
mucina però i assumim que no hem trobat cap
representació del domini ja que no ha estat descrit.
![]()
Durant tot el treball, hem intentat deduir l'estructura de la nostra seqüència
i serà l'hora de poder resumir quins han sigut les nostres troballes, i per tant,
quines són les caraterístiques de ENr123.
La seqüència Enr123 conté un elevat grau de repeticions, concretament el 40.07% ( correspon a 200364 bp) mentre que el percentatge d'illes GC on pensem que hi haurà exons és, del 36.30% . Aquesta dada ens recolza el càlcul que hem fet a l'apartat 2. Els tipus d'element repetitius i la llargària ocupada en la nostra seqüència es pot observar en la taula següent:
================================================== number of length percentage elements* occupied of sequence -------------------------------------------------- SINEs: 223 48204 bp 9.64 % ALUs 136 35299 bp 7.06 % MIRs 87 12905 bp 2.58 % LINEs: 157 85834 bp 17.17 % LINE1 76 64586 bp 12.92 % LINE2 76 20236 bp 4.05 % L3/CR1 5 1012 bp 0.20 % LTR elements: 63 43580 bp 8.72 % MaLRs 32 12165 bp 2.43 % ERVL 7 2331 bp 0.47 % ERV_classI 23 27994 bp 5.60 % ERV_classII 1 1090 bp 0.22 % DNA elements: 48 13869 bp 2.77 % MER1_type 21 4210 bp 0.84 % MER2_type 12 6240 bp 1.25 % Unclassified: 1 1563 bp 0.31 % Total interspersed repeats: 193050 bp 38.61 % Small RNA: 0 0 bp 0.00 % Satellites: 0 0 bp 0.00 % Simple repeats: 62 4238 bp 0.85 % Low complexity: 82 3096 bp 0.62 % ==================================================
Es pot observar que hi ha una gran quantitat d'elements repetitus LINEs,
sent aquests els mès abundants. En canvi, no s'han trobat evidències
de la presència de small RNA ni de micorsatèllits. El contingut de DNA de baixa
complexitat, tot i ser present, és molt poc abundant.
Després d'haver analitzat el contingut repetitiu que de la regió ENr123
( es pot visualitzar la taula en la representació de l'apartat 2 ), resumirem
quins són els dominis funcionals que hem trobat.
Dins de la regió codificant, hem pogut predir 2 gens complets que codifiquen per a
dues proteïnes anteriorment comentades. A més, tenim un gen incomplet, que
acaba en la regió que continua amb la ENr123. N'hem intentat predir
el seu domini funcional al qual codificava, tot i que els EST no ens recolzaven
la predicció .
Buscant informació sobre aquesta proteïna vam trobar que el gen que la codifica es troba al cromosoma 12 al braç 12q12. Just coincideix en la regió on es localitza Enr123, és per això que podem afirmar que aquest gen 1 codifica a aquesta proteïna.Score = 389 bits (998), Expect = e-109 Identities = 207/225 (92%), Positives = 210/225 (93%), Gaps = 9/225 (4%) Query: 1 MLLLKRQLSLDALWQELLVSSTVGAAAVSALAGGALNGVFGRRAAILLASALFTAGSAVL 60 MLLLKRQLSLDALWQELLVSSTVGAAAVSALAGGALNGVFGRRAAILLASALFTAGSAVL Sbjct: 87 MLLLKRQLSLDALWQELLVSSTVGAAAVSALAGGALNGVFGRRAAILLASALFTAGSAVL 146 Query: 61 AAANNKETLLAGRLVVGLGIGIASMTVPVYIAEVSPPNLRGRLVTINTLFITGGQFFASV 120 AAANNKETLLAGRLVVGLGIGIASMTVPVYIAEVSPPNLRGRLVTINTLFITGGQFFASV Sbjct: 147 AAANNKETLLAGRLVVGLGIGIASMTVPVYIAEVSPPNLRGRLVTINTLFITGGQFFASV 206 Query: 121 VDGAFSYLQKDGWRYMLGLAAVPAVIQFFGFLFLPESPRWLIQKGQTQKARRILSQMRGN 180 VDGAFSYLQKDGWRYMLGLA VPAVIQFFGFLFLPESPRWLIQKGQTQKARRILSQMRGN Sbjct: 207 VDGAFSYLQKDGWRYMLGLAXVPAVIQFFGFLFLPESPRWLIQKGQTQKARRILSQMRGN 266 Query: 181 QTIDEEYDSIKNNIEEEEKEVGS--------VSYPVPANKCTLRG 217 QTIDEEYDSIKNNIEEEEKEVGS +SYP P + + G Sbjct: 267 QTIDEEYDSIKNNIEEEEKEVGSAGPVICRMLSYP-PTRRALIVG 310
Function - sugar porter activity - transporter activity Process - carbohydrate transport Component - integral to membrane - membrane
Amb aquesta informació és més fàcil interpretar quina
és la funció i on es localitza.
Quan buscàvem possibles gens ortòlegs a ratolins, vam realitzar un blast on
corriem aquest proteïna contra una base de dades d'EST de ratolins. Observant aquells
resultats, podem afirmar que aquesta funció es troba molt conservada entre els dos
organismes, per la qual cosa ens fa pensar que ha actuat fortament la selecció.
Tot i conservar una estructura d'EST molt semblant, en aquesta predicció observem molta
informació i a part de mostrar els EST humans, n'hi ha d'altres. És per aquesta
raó que hem optat per no mostrar el resultat d'aquest BLAST.
La representació del domini no l'hem pogut trobar, suposem que encara no deu estar caracteritzat.Function - ATP binding - GTP binding - protein serine/threonine kinase activity - protein-tyrosine kinase activity Process - protein amino acid phosphorylation - protein transport - small GTPase mediated signal transduction
Observant la localització, tornem a verificar que es troba en la mateixa
regió que la ENr123, en el cormosoma 12 en el braç 12q12.
Assumim que els exons d'aquest gen codifiquen per a la proteïna que acabem de trobar.
Buscant l'estrucura del domini ens vam adonar que no havia estat descrit però
considerem que encara no haurà estat predit. Tampoc vam trobar en aquest cas
el GenOntology ja que la descripció d'aquesta proteïna té data de l'any
2005, és a dir, que és molt recent.
![]()
Jo, primer i per sobre de tot, voldria donar les gràcies a l'Empar, per la
paciència que ha tingut durant les últimes setmanes,
fent aquest treball amb mi.
De la mateixa manera, jo voldria agraïr la paciència que Sílvia ha
tingut per aguantar-me en els moments més estressants que he passat fent el treball.
Sense ella, el treball podria haver estat tota una pesadilla.
De part de les dues, creiem que és imprescindible, agraïr l'ajuda del Pep Abril,
ja que sense ell, i sense les hores que s'ha passat a l'aula d'informàtica amb
nosaltres, aquest treball no s'hauria pogut dur a terme. També donar gràcies a
tots els components del CRG, que ens van ajudar en els primers moments de gran
confusió.
I finalment, al suport i a l'intercanvi de coneixements bioinformàtics que
hi ha hagut entre totes les noies que feiem el treball d'anàlisi de seqüències.
Gràcies a tots.
![]()
A continuació hi ha una llista on s'especifiquen tots els servidors i webs que hem fet servir durant tot el treball :
![]()