
Maria Larroy, Paola Paoletti
4 o Biología UPF 2003-2004
1. Introducción
El genoma de Apis Mellifera (abeja común) ha sido secuenciado recientemente pero debe ser analizado para encontrar genes y otros elementos funcionales. Su secuencia genómica está disponible pero la información sobre el contenido de genes es escasa. Para obtener de forma rápida esta información se utilizan los proyectos de secuenciación de ESTs (Expressed Sequence Tag).
Los ESTs son "single-pass reads" de mRNAs expresados en células de determinados tejidos. Son secuencias cortas de entre 400-1000bp, por lo que no cubren el mRNA entero sino sólo los extremos. Los ESTs son baratos de producir y suponen un camino efectivo para obtener información sobre genes expresados. El aspecto negativo de este sistema es que al ser "single-pass reads" no siempre aseguran una buena calidad de la secuencia.
Los ESTs noveles y, en general, todas las nuevas secuencias se anotan primeramente por homología a secuencias conocidas. Utilizando BLAST o similares, estas nuevas secuencias se pueden comparar a bases de datos de secuencias con anotaciones conocidas. De este modo podemos inferir una posible función para estas nuevas secuencias.
Los ESTs de Apis Mellifera están siendo analizados utilizando bases de datos de genes conocidos en Drosophila y otros metazoos. De este análisis se ha obtenido que hay muchos ESTs de abeja que no teniendo un match en Drosophila tienen un buen match en humanos y ratón. Esto indica que hay funciones que se han perdidio en Drosophila pero que se han preservado en humano y otros vertebrados.
2. Objetivos
El objetivo de nuestro proyecto es conseguir anotar un conjunto de secuencias de ESTs de Apis Mellifera .
Analizaremos aquellos ESTs que tengan por lo menos un ORF de 450bp y BLASTX (match con algún vertebrado) y los compararemos con genes humanos de Ensembl. A partir de esta comparación eligiremos el mejor match para cada EST para poder obtener una anotación funcional. Además podremos observar posibles conservaciones funcionales entre Apis Mellifera y humano.
3. Materiales y Métodos
Datos:
| Homo Sapiens | Apis Mellifera | |
| cDNA | Contigs | |
| Clean seq. |
- cDNA: Secuencias de cDNA humano obtenidas de Ensembl.
- Contigs: ESTs solapados de Apis Mellifera con ORF de 450bp (min.) y BLASTX.
- Clean seq.: ESTs sin solapar de Apis Mellifera con ORF de 450bp (min.) y BLASTX.
Programas:
- Blastn
- Tblastx
- Parseblast.pl
- R
4. Procedimientos
Inicialmente trabajamos con las secuencias Contig de abeja ya que esperábamos obtener unos resultados óptimos, pero no ha sido así. Es por esto que nuestro proyecto consta de dos bloques; el primero, utilizando las secuencias Contig, y el segundo, utilizando las secuencias Clean.
Bloque 1: "Trabajando con Contigs"
- Obtención de las secuencias de cDNA humano de Ensembl/EnsMart .
- Obtención de las secuencias Contig de abeja de "Honey bee EST sequencing project"
- Blastn: secuencia nucleotídica query (Contig) contra la secuencia nucleotídica utilizada como base de datos (cDNA).
- Tblastx: traducción 6-frame de la secuencia nucleotídica query contra la traducció, 6-frame de la secuencia utilizada como base de datos. Hemos utilizado una E-value de 0.1, es decir, no muy restrictiva para obtener un número elevado de hits.
Los hits obtenidos con Tblastx son mayores que los obtenidos con Blastn, por lo que continuamos el estudio a partir de los resultados de Tblastx .
- Parseblast.pl: conversión del output de Tblastx a formato GFF (General Feature Format) .
- Obtención de un archivo que muestra la longitud alineada de la secuencia Contig mediante el script Contig_alignement .
- Obtención de un archivo que muestra la longitud total de la secuencia Contig mediante el script Contig_length .
- Obtención de un archivo que muestra el porcentaje de identidad entre la secuencia Contig y el cDNA humano mediante el script Identity% .
- Obtención de un archivo que muestra el %coverage de la secuencia Contig sobre el cDNA humano mediante el script Coverage% .
- Análisis de los resultados obtenidos: el coverage obtenido es muy bajo. Esto puede ser debido a que al trabajar con Contigs estamos trabajando con secuencias de ESTs solapadas y, por tanto, podemos estar perdiendo información.
Por este motivo seguimos el estudio utilizando las secuencias Clean.
Los hits obtenidos con Tblastx son mayores que los obtenidos con Blastn, por lo que continuamos el estudio a partir de los resultados de Tblastx .
Bloque 2: "Trabajando con secuencias Clean"
- Obtención de las secuencias de cDNA humano de Ensembl/EnsMart .
- Obtención de las secuencias Clean de abeja de "Honey bee EST sequencing project"
- Tblastx: traducción 6-frame de la secuencia nucleotídica query contra la traducció, 6-frame de la secuencia utilizada como base de datos. Hemos utilizado una E-value de 0.1, es decir, no muy restrictiva para obtener un número elevado de hits. Resultados de Tblastx .
No hemos corrido Blastn porque ya habíamos comprobado en el Bloque 1 que se obtienen menos hits que con Tblastx.
- Parseblast.pl: conversión del output de Tblastx a formato GFF (General Feature Format) .
- Obtención de un archivo que muestra la longitud alineada de la secuencia Clean mediante el script Clean_alignement .
- Obtención de un archivo que muestra la longitud total de la secuencia Clean mediante el script Clean_length
- Obtención de un archivo que muestra el porcentaje de identidad entre la secuencia Clean y el cDNA humano mediante el script Identity% .
- Obtención de un archivo que muestra el %coverage de la secuencia Clean sobre el cDNA humano mediante el script Coverage% .
- Elaboración de los hitogramas de coverage, tanto de los resultados obtenidos con los Contig como con los obtenidos con las secuencias Clean.
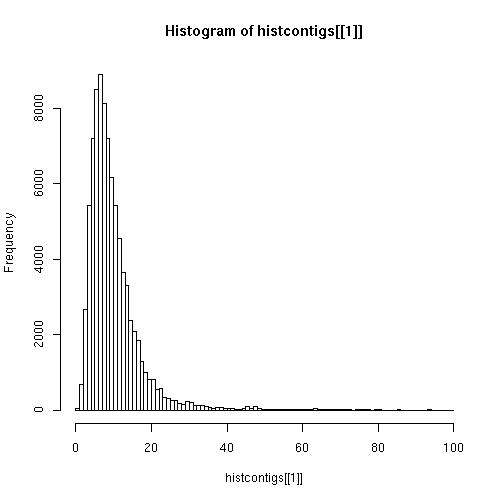 Contigs
Contigs
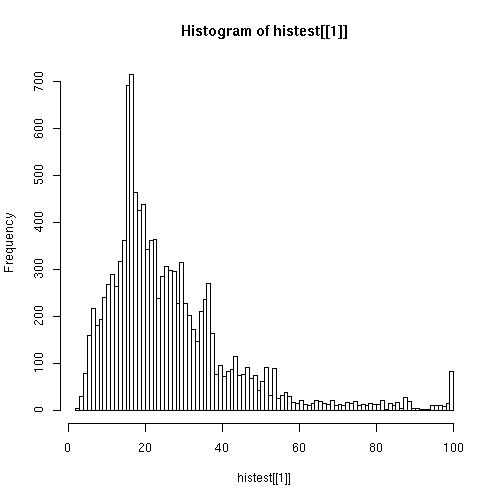 Clean
Clean
Observando los hitogramas comprobamos que los resultados de coverage son mucho mejores utilizando secuencias Clean.
- Obtención de un archivo que contiene el mejor match para cada secuencia Clean por encima de un umbral de coverage (80%) y uno de porcentaje de identidad (60%) mediante el script Threshold .
No hemos corrido Blastn porque ya habíamos comprobado en el Bloque 1 que se obtienen menos hits que con Tblastx.
Contigs
Clean
Observando los hitogramas comprobamos que los resultados de coverage son mucho mejores utilizando secuencias Clean.
5. Tratamiento de los resultados
Una vez obtenido el mejor match para cada EST, queremos anotar una función y/o descripción para cada uno de ellos.
Para esto hemos obtenido una serie de archivos con información de interés:
- Identificador del gen humano + descripción de Ensembl. click
- Identificador del gen humano + descripción de Gene Onthology. click
- Identificador del gen humano + descripción de SANBI. click
Ademá hemos desarrollado un script que nos permite comparar la información contenida en estos archivos. Los resultados son:
- Clean ESTs + descripción de Ensembl. click
- Clean ESTs + descripción de Gene Onthology. click
- Clean ESTs + descripción de SANBI. click
Los resultados obtenidos a partir de las descripciones de Gene Onthology no son completas y no aportan información. Es por esto que no las utilizamos en nuestro análisis.
Para poder obtener información significativa de estos documentos hemos realizado un archivo que resume todo lo anterior (identificador del gen + Clean EST + descripción SANBI + descripción Ensembl) mediante un script .
6. Conclusiones
Las conclusiones de nuestro proyecto las extraeremos a partir del último archivo elaborado.
Nos planteamos las siguientes cuestiones:
- ¿Encontramos algún tránscrito de Ensembl para el cual tenemos un match sin anotación identificada?
- ¿Encontramos tránscritos de Ensembl que no se expresan en cerebro?
- ¿Cuántos de los tránscritos de Ensembl se expresan en cerebro?
- En relación a las anotaciones derivadas para los ESTs, ¿cuántos ESTs se expresan en otros tejidos que no sea cerebro? ¿Cuántos tienen funciones no relacionadas con la bioquímica de cerebro?
Observando los resultados vemos que la gran mayoría de tránscritos tienen un match con una anotación identificada; tan solo dos no la tienen (BB170021B10D02 y BB170028A20B03). Para estos dos no hemos encontrado ninguna anotación, y esto puede ser debido a que se trate de genes noveles humanos que aún no han sido descritos. Únicamente para un tránscrito no hemos encontrado su lugar de expresión, sólo sabemos que se trata de una cadena de tubulina beta (BB160019A10F07).
De los que sí tienen anotación, hemos constatado que, aunque los ESTs utilizados en nuestro análisis proceden de cerebro de abeja, no todos los matches son con genes humanos expresados en cerebro. Así pues, encontramos cinco tránscritos expresados en aparato genital, tres de los cuales son de expresión neoplásica; y nueve expresados en aparato respiratorio, la totalidad de ellos presentes en neoplasia.
Estos resultados son sorprendentes porque al tratarse de ESTs de cerebro de abeja, esperaríamos encontrar que todos los matches con humano pertenecieran al sistema nervioso, pero esto no ha sido así. Una posible explicación podría ser que, al ser los ESTs secuencias cortas, alineen con tránscritos de genes humanos que no corresponden con su homólogo real.
De todos los ESTs de origen tan solo seis presentan matches con genes humanos expresados a nivel de sistema nervioso. Cuatro de estos están presentes en neoplasias y dos en cerebro:
| Gen humano | EST | Función |
| ENST00000273854.1_ENSG00000145242.3 | BB170016A20F01 | Precursor del receptor 5 de Efrina tipo A |
| ENST00000334107.1_ENSG00000145242.3 | BB170016A20F01 | Precursor del receptor 5 de Efrina tipo A |
| ENST00000263934.2_ENSG00000054523.3 | BB170029B20F04 | Proteína tumoral kinesin-like |
| ENST00000263935.2_ENSG00000054523.3 | BB170029B20F04 | Proteína tumoral kinesin-like |
| ENST00000309329.1_ENSG00000054523.3 | BB170029B20F04 | Proteína tumoral kinesin-like |
| ENST00000321943.2_ENSG00000054523.3 | BB170029B20F04 | Proteína tumoral kinesin-like |
A partir de la tabla podemos observar que se trata de dos ESTs que tienen un match con dos genes humanos que presentan diferentes tránscritos.
A modo de resumen, recogemos en esta tabla la distribución de los ESTs en los diferentes sistemas:
| S. Genital | S. Respiratorio | S. Nervioso |
| BB160007B20H05
BB160011A10D04 BB160016B10B04 BB170013A20H08 |
BB170021B10D02
BB170028A20B03 |
BB170016A20F01
BB170029B20F04 |
7. Agradecimientos
La elaboración de este proyecto no hubiera sido posible sin la valiosa ayuda de Eduardo Eyras, por su inestimable paciencia e intento de buena cara matutina al ver nuestra mano alzada; Ana Corrionero, por compartir con nosotras su sabiduría sobre links y frames; y por supuesto, a nosotras mismas, por sorprendernos cada día más.
Gracias a tod@s.