How we made it?
Materials and methodology
The aim of our study was to identify the selenoproteins in the genome of Carettochelys insculpta. Fort that, this genome was compared with the annotated selenoproteins of Mus musculus obtained from SelenoDB 1.0. The selenoproteome of Anolis carolinensis was also taken into account (obtained from SelenoDB 2.0) in order to detect genes that were not detected using the mouse.
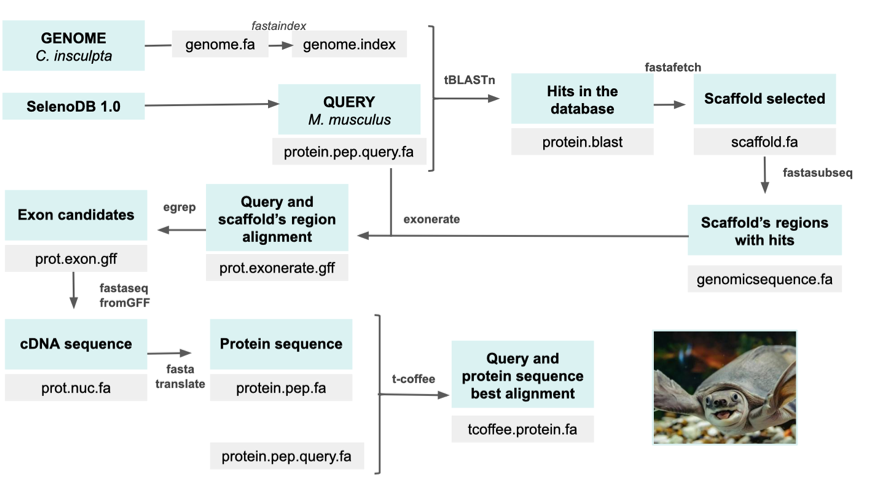
Obtaining the genome of Carettochelys insculpta
The Carettochelys genome was obtained from a public database and was provided by the teachers of Bioinformatics. In order to get it, we have to follow the next directory:
/mnt/NFS_UPF/soft/genomes/2019/Carettochelys_insculpta/genomes.fa In this directory you can find the Carettochelys insculpta genome, but to be able to use it during the selenoprotein search process it is necessary to index it (separate the genome into segments and sort it in scaffolds). The indexed genome is stored in a file called genome.index (this file was also provided to us at the beginning of this project).
Obtaining the query sequences
In order to obtain sequences of selenoproteins for Mus musculus and Anolis carolinensis, the SelenoDB (1.0 and 2.0, respectively) database has been used. From here, we obtained files called protein.pep.query.fa. Each of these files had one protein sequence of a gene of interest.
Selection of the target sequence
BLAST (Basic Local Alignment Search Tool) is a computer program for alignment of biological sequences whether they are nucleotides or amino acids. The program allows us to compare a reference sequence (query) against a large number of sequences found in a database. The program uses a heuristic algorithm and finds the sequences or hits with the higher homology between the sequences in the database and our query. It is important to mention that, since BLAST uses a heuristic algorithm, it can not be guaranteed that the perfect solution will be find as it can miss real hits that do not show much similarity.
In our case, tBLASTn has been used because we were interested in the alignment of a protein sequence (query) with a database of nucleotide sequences (the Carettochelys insculpta genome).
To execute the program the following command was used:
tblastn-query protein.pep.query.fa. -db /mnt/NFS_UPF/soft/genomes/2019/Carettochelys_insculpta/genome.fa-outfmt 6 -out protein.blast -evalue 0.001 The output of tBLASTn contains a list of the possible alignments between the query and the genome against which it has been aligned. Each of these alignments is a hit with an E-value lower than 0.001, which allows us to choose only significant hits. This parameter describes the number of hits, which is, the number of alignments that can only be expected by randomization through BLAST analysis.
It should be noted that in the case of multiple hits for a query protein, percentage of coverage was calculated manually in order to discard those scaffolds with less than 60% of query coverage. For these scaffolds, we stopped their analysis here. At this point, Anolis carolinensis was taken as a reference and the study started again. In the cases where the coverage continued being less than 60%, the analysis of the protein stopped here, and we considered that the respective gene is missing in Carettochelys insculpta genome.
To summarize, the sequences of Mus musculus (or Anolis carolinensis when no result was obtained with the previous species) proteins have been aligned to Carettochelys insculpta genome to find out possible regions with the respective coding genes. This was performed by tBLASTn and we obtained a file called protein.blast.
Extraction of the genomic region
Once all the hits have been obtained, the genomic region (in which the alignment showed that we could possibly find the gene) must be extracted. Since the genome was already indexed, the Fastafetch program could be executed directly, which allows the extraction of the scaffold in which we can find the selected hit. To execute this program the following command was used:
fastafetch /mnt/NFS_UPF/soft/genomes/2019/Carettochelys_insculpta/genome.fa/mnt/NFS_UPF/soft/genomes/2019/Carettochelys_insculpta/genome.index SCAFFOLD > scaffold.fa Where “SCAFFOLD” refers to the name of the selected scaffold and scaffold.fa was the name that we gave to its output.
Next, within the selected scaffold, the region of interest should be further delimited in order to achieve shorter and more accurate sequences of the fragment that possibly contains the selenoprotein-coding gene. With the aim of extracting the whole gene of interest, the positions of the hit obtained in the BLAST are taken and the downstream and upstream margins of the alignment are expanded. The size of the expansion that was carried out was 50,000bp upstream and 50,000bp downstream nucleotides. By doing this, it is also guaranteed to include the SECIS element of the gene. However, this size may vary depending on the length and properties of the query or the scaffold. In order to narrow down the region, the Fastasubseq program is used by the following command:
fastasubseq scaffold.fa start lenght > genomicsequence.fa In this command: the selected scaffold (scaffold..fa), the nucleotide where our region starts (start) and the length of nucleotides that will contain this (lenght) were introduced. The selected region will be stored in an output file called genomicsequence.fa in which "sequence" refers to the name of the selenoprotein that was being analyzed.
Prediction of the gene
Once the DNA fragment containing the hit of interest has been extracted through the Fastasubseq, it is necessary to ensure that the region codifies for a protein, and get the region where it is coded. Exonerate is a program that allows to predict the gene structure of the problem sequence of interest in the Carettochelys insculpta genome. The command to run this program is the following:
exonerate -m p2g --showtargetgff -q protein.pep.query.fa -t genomicsequence.fa -n 1 > prot.exonerate.gff where -m indicates the alignment model, in this case p2g (protein to genome), that is, compare the query against the genome. The -q parameter indicates where the file is found while the -t we indicates the sequence against which the query is compared, that is, the scaffold region in which the gene of interest should be. Also, -n option reduces the amount of output generated, it reports the best N results for each query. Finally, prot.exonerate.gff stores the output file in GFF format.
When we want to execute the command, it is important to change the possible "U" for "X" in the file where the query is, since Exonerate does not recognize the U letter (Sec).
In order to get only the exons, the egrep -w exon command was used:
egrep -w exon prot.exonerate.gff > prot.exon.gff Next, the FastaseqfromGFF.pl program has been used, which will extract the exon sequence in FASTA format by doing:
fastaseqfromGFF.pl genomicsequence.fa prot.exon.gff > prot.nuc.fa where genomicsequence.fa is the subsequence that was extracted previously, prot.exon.gff is the file that contains only exons and prot.nuc.fa is the output file that will contain the DNA retrieved from the genome in FASTA format.
Finally, once the DNA retrieved from the genome has been obtained in FASTA format, the Fastatranslate program has been executed to obtain the amino acid sequence:
fastatranslate -f prot.nuc.fa -F 1 > protein.pep.fa where prot.nuc.fa is the subset that contains the DNA retrieved from the genome in FASTA format, the argument -F 1 is used to limit the selected reading frame, in this case, it is the first one offered by the program, and finally, protein.pep.fa is the output file that will contain the amino acid sequence.
T-coffe
T-COFFEE (Tree-Based Consistency Objective Function for Alignment Evaluation) is a program that allows us to do a global alignment of the resulting protein obtained from the Carettochelys insculpta genome, with the query protein used in each case. The result, therefore, allowed to evaluate the homology between the query and the predicted sequence. The T-COFFEE has been executed with the following order:
t_coffee protein.pep.fa protein.pep.query.fa > tcoffee.protein.fa where protein.pep.query.fa is the query; protein.pep.fa, the sequence obtained from the fastatranslate and tcoffeeprotein.fa is the file that contains the alignment between the predicted sequence and the query.
Automation
All the pipeline was automatized from the tBLASTn results until the T-coffee through the following Python program. However, some proteins that showed hits in the same scaffold but with a huge distance between them, were analyzed separately by hand. Moreover, the calculation of the % of coverage and phylogenetic analyses were made manually.
Phylogenetic analysis
In the case of query proteins with multiple possible coding-genes in Carettochelys insculpta genome (multiple regions retrieved from tblastn, resulting in multiple predicted proteins), a phylogenetic analysis was performed to check if all of them really corresponded to the protein of interest (duplication), or if they resulted from alignments of the query protein in genes coding for proteins of the same family. This analysis was particularly important for the protein families. For that, phylogeny.fr was used using the “one click” option. Briefly, the predicted proteins for a given family were aligned with the respective query proteins using Muscle and the phylogenetic tree was reconstructed with PhyML. This analysis allowed us to identify the most similar predicted protein to our query reference and, therefore, which one should be further analyzed (see specific cases in discussion). As mentioned before, we could also identify duplicated and deleted genes.
Seblastian
Once the global alignment from T-coffee was obtained, the Seblastian program was used to predict SECIS elements (Senelocysteine Insertion Sequence) and the selenoproteins.
As explained in the introduction (see introduction), SECIS elements are necessary in protein translation and, therefore, they confirm the predictions made from T-coffee.
This search was made with sequences obtained from fastasubseq. It is important to observe in which strand the gene was located: forward or reverse, because the SECIS element should appear on the 3’ end of the predicted gene, not more than 5.000 nucleotide positions away from the first exon of the protein. Those SECIS predictions that appeared in the opposite strand or did not accomplished the criteria explained before, were discarded.