MATERIALS I MÈTODES
Aquest projecte es basa en localitzar i anotar les selenoproteïnes que es troben en el genoma de l’espècie Callorhinus ursinus, així com la maquinària necessària per a la seva síntesis i funcionament. Per trobar les regions codificants per aquestes proteïnes vam agafar de referència l’anotació de les selenoproteïnes d’una espècie propera a la d’interès, obtinguda al SelenoDB. En aquest cas, l’espècie filogenèticament més propera a Callorhinus ursinus, era el Canis lupus, però ens vam adonar que les seves selenoproteïnes no estaven gaire conservades, pel que vam decidir agafar com a referència el genoma de l’humà (Homo sapiens). Tot i que sabem que aquesta espècie és llunyana a la nostra és l’espècie que està millor caracteritzada i amb les selenoproteïnes més ben anotades.
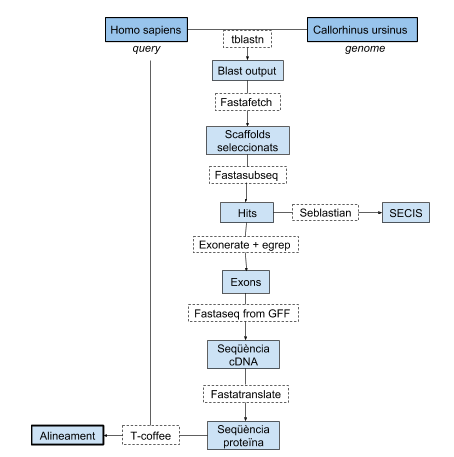
Per tal d’obtenir les queries de les diferents selenoproteïnes, vam descarregar les seqüències a partir de la base de dades SelenoDB i es van utilitzar com a queries. Cada una d’aquestes va ser guardada en un document de text en format FASTA i va ser analitzada posteriorment. Totes les queries s’han guardat en una carpeta per tenir-les ben organitzades i poder-les utilitzar al executar el programa.
Seguidament, vam eliminar manualment alguns símbols (#,%,@) que apareixien en les seqüències mitjançant l’editor de text EMACS. En cas de que no els haguessim eliminat, els resultats obtinguts dels programes serien incorrectes.
El genoma de Callorhinus ursinus ens l’ha proporcionat el professorat. Es pot obtenir en el següent enllaç:
/mnt/NFS_UPF/bioinfo/BI/genomes/2018/Callorhinus_ursinus/genome.fa
Per tal de realitzar la predicció vam crear un programa que automatitzés aquest procés de manera que agilitzés l’obtenció dels resultats. El llenguatge que s’ha utilitzat és el “bash”. Aquest programa es pot trobar en el següent enllaç:
Tot i així, vam realitzar un altre programa per analitzar de forma individual, per una banda aquelles selenoproteïnes de les quals l’score obtingut en el T-Coffe no era l’idoni; i per l’altre, aquelles que pertenyien a les famílies de Gpx, DI i TR. Al executar aquest programa s’ha d’especificar de manera manual el nom de l’Scaffold i l’inici i el final de la posició on es troba dins del genoma de Callorhinus ursinus.
Una vegada hem obtingut els queries, substituïm les “U” per “X”, ja que aquest símbol ens indica l’aminoàcid de selenocisteïna que es troba a les proteïnes en qüestió. Aquest és imprescindible ja que els programes que executem no reconeixen el caràcter “U”. Vam fer-ho manualment amb la següent comanda:
sed ‘s/U/X/g’ proteina.fa > proteina.fa
Per tenir permís per poder executar el nostre programa hem d’utilitzar la següent comanda:
chmod u+x programa.sh
A continuació explicarem els passos que hem seguit per crear el nostre programa:
EXPORTS
Dins del programa hem inclòs l’export que necessitem per poder treballar amb el FastaseqfromGFF.pl.
export PATH=/mnt/NFS_UPF/bioinfo/BI/bin:$PATH
PATHS
També s’inclouen els diferents paths on volem que es guardin els fitxers:
path_files="/mnt/NFS_UPF/bioinfo/BI/genomes/2018/Callorhinus_ursinus"
path_db="$path_files/genome.fa"
path_out="./blast"
path_proteins="./prots"
path_scaffolds="./scaffolds"
path_subseq="./subseqs"
path_exonerate="./exonerate"
path_GFF="./GFF"
path_secis="./secis"
path_tcoffee="./tcoffee
El primer que s’executa és el BLAST (Basic Local Alignment Search Tool), un programa que serveix per comparar cada query amb el genoma de Callorhinus ursinus. Es basa en un algorisme heurístic que compara les seqüències de DNA i selecciona els hits més significatius. En el nostre cas utilitzem el tBLASTn amb el qual obtindrem les seqüències d’homologia que existeixen entre les selenoproteïnes (Homo Sapiens) i el genoma de Callorhinus ursinus. Per evaluar la importància de cada hit hem tingut en compte l’e-value. En el programa hem posat l’argument -evalue 0.001, la qual permetrà que només ens mostri el hits que són estadísticament significatius (els que tenen un evalue inferior a 0.001). A més, també hem afegit l’argument -outfmt que ens donarà el resultat del Blast en forma de taula. La comanda és la següent:
tblastn -query $path_proteins/$prot -db $path_db -evalue 0.001 -outfmt 6 -out $path_out/$prot.blast
Després de realitzar el BLAST hem executat el Fastafetch, el qual ens permet extreure les regions d’interès, és a dir, els scaffolds. Aquestes regions del genoma de Callorhinus ursinus concorden amb les selenoproteïnes del genoma humà. En el cas de les proteïnes que no hem analitzat manualment, hem declarat la variable scaffold, de manera que agafi l’scaffold amb l’e-value més petit:
scaffold=$(head -n 1 $path_out/$proteina.blast | cut -f2)
La comanda que hem utilitzat és la següent:
fastafetch $path_db $path_files/genome.index $scaffold >
"$path_scaffolds/$proteina.fastafetch"
L'índex del genoma que hem utilitzat (genome.index) també el vam trobar a la carpeta que ens va proporcionar el professorat.
Seguidament, executem el Fastasubseq que ens permet extreure les regions específiques de cada hit que es troben en els diferents scaffolds. Per poder afirmar que la nostra seqüència conté el gen d’interès l’hem d’allargar 50.000 nucleòtids tant en la direcció 3’ (downstream) com en la 5’ (upstream). A més, també hem inclòs una condició que ens digués si la seqüència és forward o reverse.
if (($inici>$final)); then
inici1=$final
final1=$inici
echo "la seqüència és reversa"
else
inici1=$inici
final1=$final
echo "la seqüència és forward"
fi
La comanda és la següent:
inici2=$(($inici1-50000))
final2=$(($final1+50000))
length=$(($final2-$inici2))
fastasubseq $path_scaffolds/$proteina.fastafetch $start $lengthfinal > "$path_subseq/$proteina.subseq"
Aquest programa permet predir el gen d’interès mitjançant la comparació entre la regió obtinguda amb el Fastasubseq (expressada en nucleòtids) i la seqüència de la proteïna humana (expressada en aminoàcids).
exonerate -m p2g --showtargetgff -q $path_proteins/$proteina -t
$path_subseq/$proteina.subseq > "$path_exonerate/$proteina.exonerate.gff
La “m” ens indica el tipus d’alineament, és a dir, protein to gene (p2g); la “q” és la query; la “t” és la regió resultant del fastasubseq; i el “showtargetgff” indica que el fitxer de sortida serà en format gff.
A més, amb l’argument -egrep podem obtenir els diferents exons que codifiquen per les selenoproteïnes:
egrep -w exon $path_exonerate/$proteina.exonerate.gff > "$path_exonerate/$proteina.exon.gff"
Donat que l’Exonerate ens dóna com a resultat la seqüència de cDNA en format GFF, hem d’utilitzat la següent comada per obtenir-lo en format FASTA:
fastaseqfromGFF.pl $path_subseq/$proteina.subseq $path_exonerate/$proteina.exon.gff >
"$path_GFF/$proteina.pred.nuc.fa"
Per tant, el que s’obté és el cDNA que codifica per la proteïna que hem predit.
El següent pas és traduir el cDNA a aminoàcids, obtenint així la proteïna.
fastatranslate -F 1 $path_GFF/$proteina.pred.nuc.fa > "$path_GFF/$proteina.pred.aa.fa"
El problema és que el fitxer que s’obté del FASTAtranslate converteix totes les X que haviem posat inicialment, és a dir, les selenoproteïnes predites, per un “*”. Això causa un error a l’hora d’executar el T-coffe, per lo que hem fer una comanda per canviar els “*” per “X”:
sed 's/*/X/g' $path_GFF/$proteina.pred.aa.fa > $path_GFF/$proteina.pred.aa.be.fa
Finalment executem el programa T-coffe que fa un alineament global entre les seqüències obtingudes de la base de dades SelenoDB i la proteïna predita de la nostra espècie. D’aquesta manera podrem obtindre l’homologia entre les dues seqüències i l’score de l’alineament.
t_coffee $path_GFF/$proteina.pred.aa.fa $path_proteins/$proteina > "$path_tcoffee/$proteina.tcoffee"
Per confirmar la presencia de selenoproteïnes hem de predir els elements SECIS ja que son essencials per què l’mRNA presenti una selenocisteïna en la seva cadena. Si aquest element no estigués, el codó UGA correspondria a un codó STOP, deixant de ser una selenoproteïna. Aquesta predicció l’hem fet gràcies al programa SEBLASTIAN i en alguns casos també el SECISearch3 per acabar de confirmar.
Per últim, hem realitzat un arbre filogenètic per les tres famílies de proteïnes més complexes (TR, GPx i DI) per assegurar-nos de que la proteïna predita estigués propera a la query. Per fer-ho hem utilitzat la web de Phylogeny.fr que es dedica a representar i analitzar relacions filogenètiques entre seqüències moleculars. Hem hagut d’obtenir en format multifasta totes les proteïnes de les tres famílies diferents. Això ho hem insertat com a input a la web i aquesta ens ha proporcionat directament els arbres filogenètics.