L’objectiu d’aquest estudi és predir i anotar les selenoproteïnes del Xiphophorus helerii i determinar les seves proteïnes de maquinària. Per dur a terme aquest treball hem comparat el genoma del X. hellerii amb el genoma del Danio rerio (també conegut amb el nom de Zebrafish) ja que és un peix completament anotat degut a la seva importància com a model de recerca i a més, és filogenèticament proper a la nostra espècie. Hem utilitzat el seu genoma detallat per comparar les seves seqüències de selenoproteïnes amb el genoma de Xiphophurus hellerii.
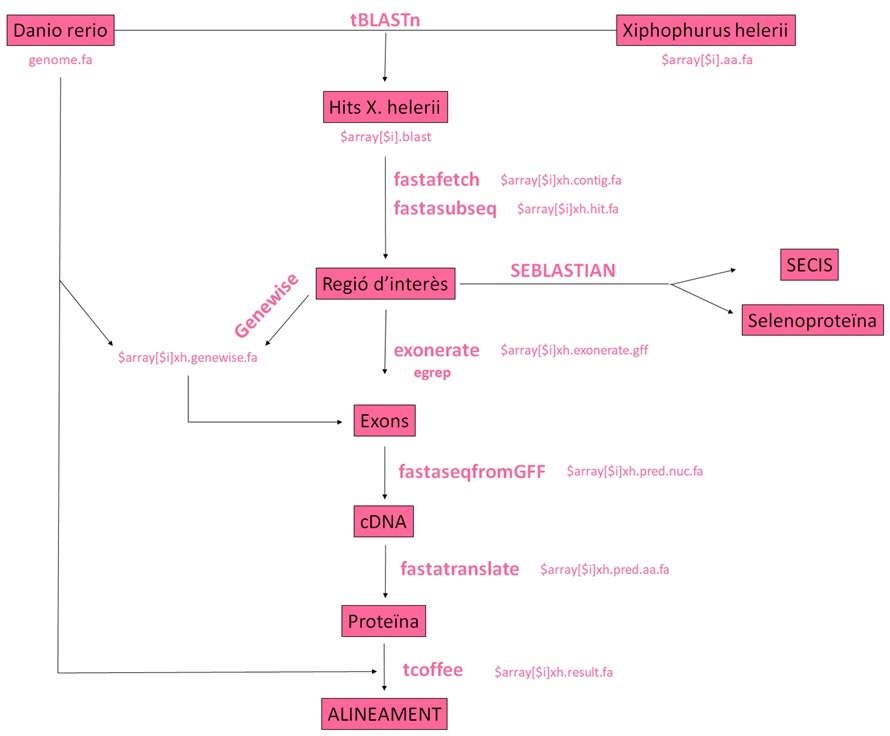
A partir de les selenoproteïnes del Danio rerio (genoma de referència: genome.fa) hem definit mitjançant el tBLASTn les regions amb alta homologia amb les selenoproteïnes, els homòlegs amb cisteïna i les proteïnes de maquinària del Xiphophurus hellerii. Trobats els hits, hem corregut el fastafetch i el fastasubseq per extreure i seleccionar les regions dels scaffolds que contenen un hit. Un cop obtingudes les regions d’interès, hem corregut una sèrie de programes de predicció (Exonerate, Genewise, Seblastian) per extreure els els exons i predir si contenien elements SECIS i/o estructures 3D corresponents a selenoproteïnes a l’mRNA. Posteriorment, hem corregut el fastaseqfromGFF per obtenir el cDNA i el fastatranslate per traduir-ho a proteïna. Finalment, hem utilitzat el T-Coffee per comparar les queries del genoma del Zebrafish mitjançant un alineament amb les nostres prediccions i poder valorar la qualitat de l’alineament i conseqüentment de la nostra predicció.
Selecció del genoma
Els nostres professors ens van proporcionar el genoma del Xiphophurus helerii i nosaltres ens l’hem descarregat de la següent font:
/mnt/NFS_UPF/bioinfo/BI/genomes/2018/Xiphophorus_hellerii/genome.fa
Obtenció de les queries
Les “queries” són el genoma de referència (D. rerio) amb el qual estem comparant el X. hellerii per predir les seves selenoproteïnes. Hem obtingut les selenoproteïnes del Zebrafish (genoma de referència) de la web SelenoDB i ens les hem descarregat amb el nom “$array[$i].aa.fa”, on $array[$i] correspon a cada selenoproteïna del Zebrafish.
Degut a que algunes famílies de proteïnes del Danio rerio no presentaven la subfamília especificada, les hem renombrat amb el nom seguit d’un nombre. Per exemple, la família de selenoproteïnes W (SELENOW) presenta tres proteïnes anotades com "NONE" i les vam renombrar com SELENOW1, SELENOW2 i SELENOW3.
Pel que fa a les proteïnes homòlogues en cisteïna, hem inclòs únicament les proteïnes més llargues degut a que al ser paràlogues hem considerat que serien les que tindrien més possibilitats de donar-nos hits significatius.
Llista de selenoproteïnes del Zebrafish utilitzades: sel15, eEFsec, SELENOE, GPx1a, GPx1b, GPx2, GPx3a, GPx3b, GPx4a, GPx4b, GPx7, GPx8, DIO1, DIO2, DIO3a, DIO3b, MsrA, PSTK, SBP2, SecS, SEPHS2, SELENOH, SELENOI, SELENOJ, SELENOK, SELENOL, SELENOM, SELENON, SELENOO1, SELENOO2, SELENOP1, SELENOP2, MSRB1a, MSRB1b, MSRB2, MSRB3, SELENOS, SELENOT1, SELENOT1b, SELENOT2, SELENOU1a, SELENOU2, SELENOU3, SELENOW1, SELENOW2, SELENOW3, TXNRD2, TXNRD3, SECp43.
Substitució de U per X
Com que programes com l’Exonerate o el T-Coffee solen llegir malament l’aminoàcid de la selenocisteïna (U) que tenen les selenoproteïnes, l’hem substituït per una X. A més, com que els fitxers fasta extrets del SelenoDB també contenien símbols com per exemple @, # o % que podien ser malinterpretats, hem decidit eliminar-los corrent les següents comandes:
$sed -i 's/U/X/g' inputs.aa/$array[$i].aa.fa
$sed -i 's/[#@%]//g' inputs.aa.fa/$array[$i].aa.fa
Procés de predicció
Hem automatitzat l’obtenció dels resultats mitjançant un programa de Perl que ens genera automàticament les prediccions de les selenoproteïnes. Per accedir a la pipeline del programa feu clic aquí.
Posteriorment, hem fet l’anàlisi de les prediccions així com els càlculs quantitatius del nombre de mutacions en cada alineament i les diferències de longitud de cada seqüència.
Tot i automatitzar tots els passos, hem comprovat manualment les proteïnes que no alineaven correctament (scores baixos) i hem realitzat canvis en la pipeline per analitzar diferents scaffolds.
Finalment, hem inclòs a l’anàlisi de les proteïnes el nombre d’aminoàcids que canvien respecte a la query. Per automatitzar el càlcul per a totes les proteïnes hem creat un programa de Perl que l’hem anomenat numcanvisaa.pl que ens comptabilitza la longitud de la seqüència predita, el nombre d’identitat (coincidències) entre les seqüències i finalment el percentatge d’aminoàcids modificats. D’aquesta manera, depenent el grau d’identitat entre ambdós seqüències podem inferir en la seva homologia.
tBLASTn
Hem utilitzat el programa BLAST per comparar cada query d’aminoàcids del Dario rerio ($array[$i]) amb el genoma prèviament indexat del Xiphophurus hellerii, que és la nostra base de dades (-db).
tblastn -evalue 0.005 -query input.aa/$array[$i].aa.fa (-db) /mnt/NFS_UPF/bioinfo/BI/genomes/2018/Xiphophorus_hellerii/genome.fa -out output.blast/$array[$i].blast -outfmt 6
Per avaluar la significància de cada hit hem fixat un E-value -evalue de 0,005 per descartar els hits no significants (en cas que n’hagués). L’E-value descriu quantes vegades esperem trobar aleatòriament un alineament tant bo com el que tenim a la nostra base de dades, per tant, no acceptarem E-values majors que 0,005.Hem definit el format de sortida (-outfmt 6) per especificar com volem que el programa ens mostri alineament: només ens mostrarà els hits.
El fitxer de sortida generat conté els següents elements per cada hit:
- Contig: correspon al fragment del genoma on el hit ha sigut trobat.
- Start: posició inicial del hit.
- End: posició final del hit.
- E-value: mesura de la significà del hit.
Fasta fetch
Després de la selecció automàtica dels contigs significants per cada proteïna, hem creat un fitxer amb la seqüència del scaffold que conté el hit.
fastafetch /mnt/NFS_UPF/bioinfo/BI/genomes/2018/Xiphophorus_hellerii/genome.fa /mnt/NFS_UPF/bioinfo/BI/genomes/2018/Xiphophorus_hellerii/genome.index $scaffold > output.fastafetch/$array[$i]xh.contig.fa
Fasta subseq
Extenem la búsqueda 50.000 nucleòtids tant per 5’ com per 3’ per no perdre part de la seqüència a causa dels introns i extraiem les coordenades inicials i finals de regions que contenen el hit que ha sigut alineat pel BLAST.
fastaseqfromGFF.pl output.fastasubseq/$array[$i]xh.hit.fa output.exonerate/$array[$i]xh.exonerate.gff > output.fastaseqfromgff/$array[$i]xh.pred.nuc.fa
Exonerate
Seleccionem únicament els exons de la regió d’interès i els separem dels introns amb la funció egrep.
exonerate -m p2g --showtargetgff -q input.aa/$array[$i].aa.fa -t output.fastasubseq/$array[$i]xh.hit.fa -n 1 | egrep -w exon > output.exonerate/$array[$i]xh.exonerate.gff
- q: query → seqüència d’aminoàcids de la proteïna del peix zebra que hem tret del SelenoDB.
- t: target → regió que ens interessa del genoma del X. hellerii.
- m:utilitzarà el model p2g per comparar la proteïna amb el genome.
- n:indica que agafem únicament la primera fila
Genewise
Hem contrastat les homologies obtingudes per l’exonerate per comprovar que ens havia predit bé les proteïnes. Es poden trobar tots els fitxers aquí.
genewise -pep -pretty -cdna -gff input.aa/$array[$i].aa.fa output.fastasubseq/$array[$i]xh.hit.fa > output.genewise/$array[$i]xh.genewise.fa
- cdna: mostra la seqüència predita de cDNA alineada.
- pep:mostra el pèptid predit.
- pretty:mostra l’alineament.
- gff:defineix el format de sortida en gff.
- both:considera ambdues cadenes: la forward i la reverse.
FastaseqfromGFF
Basant-nos amb els resultats de l’exonerate, obtenim la seqüència de cDNA que codificarà per la proteïna predita.
Basant-nos amb els resultats de l’exonerate, obtenim la seqüència de cDNA que codificarà per la proteïna predita.
Fastatranslate
Traduïm el cDNA per obtenir l’estructura primèria de la nostra proteïna predita.
fastatranslate -f output.fastaseqfromgff/$array[$i]xh.pred.nuc.fa -F 1 > output.fastatranslate/$array[$i]xh.pred.aa.fa
- f: fasta input file, és a dir, el que hem tret del fastaseqfromgff. Indica que el fitxer conté únicament exons en format nucleotídic.
- F:indica el marc de lectura (3 forward i 3 reverse). Sempre indiquem l’1.
Substitució de * per X
Canviem els * corresponents a les selenocisteïnes predites pel fastatranslate per X ja que sinó el T-Coffee els alinea amb un gap a la posició anterior enlloc de amb la selenocisteïna.
system ("sed -i 's/*/X/g' output.fastatranslate/$array[$i]xh.pred.aa.fa");
T-Coffee (Tree-based Consistency Objective Function for alignment Evaluation)
Comparem les queries del D. rerio amb les proteïnes predites del X. hellerii mitjançant l’elaboració d’un alineament global, que ens donarà informació sobre la seva homologia.
t_coffee input.aa/$array[$i].aa.fa output.fastatranslate/$array[$i]xh.pred.aa.fa > output.tcoffee/$array[$i]xh.result.fa 2>/dev/null
SECIS i Seblastian
Els elements SECIS són els responsables de la síntesi del codó de la selenocisteïna a partir del codó UGA (que usualment és un codó STOP). Mitjançant la localització d’aquests elements, hem confirmat la predicció de les selenoproteïnes.
Hem utilitzat el programa Seblastian per predir, a partir del fastasubseq, els elements SECIS localitzats a l’extrem 3’UTR que es troben en el mateix marc de lectura. En última instància confirmar la presència de selenoproteïnes en la nostra predicció.
Arbres filogenètics
Finalment, hem utilitzat la web phylogeny.fr. per crear un arbre filogenètic de les famílies de proteïnes més complexes per comprovar que els alineaments eren bons i per tant, verificar que cada predicció (l’output del fastatranslate) s’alineava amb la seva homòloga (les queries del D. rerio).