MATERIALES Y MÉTODOS
El objetivo de este estudio es identificar las selenoproteínas, los homólogos de cisteína y las proteínas de maquinaria presentes en el genoma de Eidolon helvum. Para ello hemos utilizado técnicas in silico, es decir, programas informáticos que nos permiten realizar alineamientos basados en homología de secuencias. Para poder explicar detalladamente todo el proceso vamos a dividir los resultados en diferentes apartados, cada uno con sus correspondientes programas:
OBTENCIÓN DEL GENOMA PROBLEMA
El genoma de Eidolon helvum fue secuenciado por la School of Biological & Chemical Sciences (Queen Mary University of London), que podemos encontrar en la siguiente dirección (GenBank ID: AWHC00000000.1).
Para la realización del trabajo, el genoma completo nos fue facilitado por los profesores de la asignatura. Podemos acceder a él a través del siguiente directorio:
Los profesores también nos facilitaron el genoma indexado, que deberemos utilizar más adelante para escoger un scaffold y determinar la presencia de las selenoproteínas en nuestro genoma. Esta es la dirección del directorio:
OBTENCIóN DE LAS QUERYS
Todas las selenoproteínas las obtuvimos de la base de datos SelenoDB. Cuando fue posible, utilizamos como query aquellas selenoproteínas que estaban bien anotadas en la especie Pteropus vampyrus, ya que es la especie filogenéticamente más cercana a Eidolon helvum que presenta un genoma secuenciado y sus selenoproteínas anotadas.
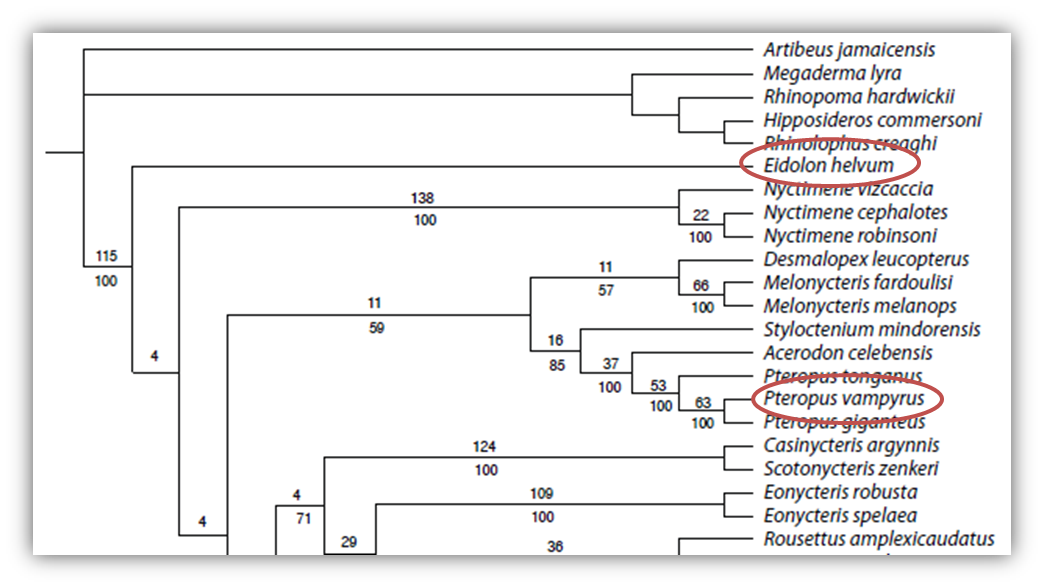
Aun así, había algunas selenoproteínas de P. vampyrus que su anotación no era correcta o que simplemente no estaban presentes en esta especie. En estos casos, recurrimos a las selenoproteínas anotadas en el genoma humano (Homo sapiens), ya que aunque filogenéticamente es una especie mucho más lejana, contiene un genoma anotado de gran calidad y sus selenoproteínas estan bien anotadas.
Cabe destacar que para poder realizar la anotación en nuestro genoma problema tuvimos que cambiar los aminoácidos de selenocisteína (U) por el carácter X, ya que los programas utilizados no son capaces de reconocer este símbolo.
Para poder realizar el análisis necesitamos una serie de programas esenciales que deben ser exportados previamente a la búsqueda de selenoproteínas. Como se explicará más adelante hemos automatizado el proceso para no tener que copiar cada vez los siguientes comandos:
| $ export PATH=/cursos/BI/bin:$PATH | fastaseqfromGFF.pl |
| $ export PATH=/cursos/BI/bin/ncbiblast/bin:$PATH | NCBI Blast |
| $ cp /cursos/BI/bin/ncbiblast/.ncbirc ~/ | NCBI Blast |
| $ export PATH=/cursos/BI/soft/exonerate/i386/bin:$PATH | Exonerate |
| $ export PATH=/cursos/BI/soft/t_coffee/i386/bin:$PATH | t_coffee |
| $ export PATH=/cursos/BI/soft/genewise/i386/bin:$PATH | GeneWise |
| $ export WISECONFIGDIR=/cursos/BI/soft/genewise/i386/wise2.2.0/wisecfg/ | GeneWise |
![]()
BLAST
La herramienta BLAST (Basic Local Alignment Search Tool) es el programa más usado para comparar información procedente de secuencias biológicas primarias, como ácidos nucleicos o DNA. Su base es un algoritmo heurístico que alinea localmente, en búsqueda de similitud, una secuencia problema con secuencias provenientes de una base de datos. Hay más información disponible al respecto en la web oficial de BLAST.
Existen diferentes versiones de esta herramienta en función de los tipos de secuencias que se quieran alinear. En nuestro caso estamos analizando una secuencia de aminoácidos como query, por lo que hemos utilizado t-blastn, modalidad que nos permite comparar nuestra query inicial proteica con una base de datos de nucleótidos.
Como resultado obtendremos los alineamientos entre query y nuestra base de datos (genoma) con sus respectivos valores de e-value, similitud, identidad, etc. A menor e-value habrá menos posibilidades de que ese alineamiento se haya dado por azar, lo que dará mayor credibilidad al resultado. En nuestro caso solo se han analizado aquellos alineamientos con un e-value menor de 0.0001. Estos son los comandos necesarios para emplear el programa:
En nuestro programa hemos utilizamos un comando ligeramente distinto, con el objetivo de transformar el output y así emplearlo para seleccionar el scaffold de interés más adelante. Este es el comando que hemos utilizado:
Estos son los argumentos utilizados en nuestro análisis y su función:
- -p: especifica el tipo de búsqueda.
- -i: especifica el input (la query)
- -d: especifica la base de datos.
- -e: especifica el límite del e-value admitido, no se admitirán valores mayores.
- -m: especifica la visualización del resultado. El valor 8 es para es para dar un formato tabular que utilizaremos luego.
- -o: especifica el formato de los resultados.
Hay que destacar que BLAST tiene como contrapartida el hecho de que puede conllevar la pérdida de alineamientos reales pero que no presentan una similitud muy elevada. Por esta razón hay que utilizar otros programas complementarios para validar la predicción.
![]()
Extracción del scaffold de interés
Un vez obtenemos el resultado del blast debemos determinar cual es la región que contiene nuestro hit para utilizarla más adelante en el análisis. Para ello utilizaremos una serie de herramientas que nos permitirán acotar la región.
Fastaindex o extracción de los scaffolds más significativos
Como se ha explicado anteriormente el índice de nuestro genoma nos fue proporcionado por los profesores. En el caso de que fuera necesario generar un índice nuevo a partir del genoma inicial utilizaríamos el siguiente comando:
fastafetch o extracción de los scaffolds más significativos
Mediante la herramienta fastafetch podemos extraer los scaffolds de los hits más significativos, para ello es condición necesaria tener el índice creado. Como identificador utilizamos el nombre del scaffold que presente un resultado significativo, el programa lo buscará en el genoma indexado para extraer toda la longitud del scaffold y guardarla en un nuevo archivo. Para llamar al programa utilizamos el siguiente comando:
Fastasubseq o extracción de la región donde se encuentra nuestro gen
Una vez que tenemos el scaffold de interés necesitamos acotar la región de alrededor de nuestro hit donde podemos encontrar nuestro gen. Se utilizan las posiciones obtenidas en el BLAST y se expanden los límites de manera que englobemos a todo el gen, tanto upstream como downstream.
En nuestro caso hemos decidido ampliar esta región 25000 nucleótidos downstream del inicio de nuestro blast y otros 25000 upstream del final de este. Este es el comado que hemos utilizado:
Particularmente, por nuestro genoma, podemos tener problemas al ampliar estos 50000 nucleótidos totales ya que podríamos superar la longitud total del scaffold y producir un error. Para solucionar el problema hemos utilizado una herramienta que nos permite determinar el límite de nuestro scaffold y así evitar un error de ejecución.
Esta herrramienta es fastalength y la utilzamos con el siguiente comando en nuestro programa:
![]()
Predicción de gen
Exonerate
Exonerate constituye una herramienta muy completa para realizar pairwise sequence comparison. Exonerate además permite generar una anotación del gen con el número de intrones y exones mediante el alineamiento de la query con la región genómica seleccionada. En nuestro caso la secuencia es la región de DNA extraída previemante con fastasubseq.
Para ejecutar el programa utilizamos el siguiente comando:
Exonerate puede utilizar diversos argumentos, estos son los que hemos utilizado:
- -m: especifica el tipo de alineamiento, p2g (protein to genome).
- --showtargetgff: determina el output como GFF.
- -q: especifica la query como formato fasta.
- -t: especifica que la secuencia target está en formato fasta.
Una vez tenemos la información podemos extraer la secuencia de los exones y guardarla mediante:
Para aislar los diferentes exones utilizamos el siguiente comando:
El archivo contendrá la secuencia de cDNA correspodiente a los exones en la proteína predicha. Para poder traducirlo a nucleótidos necesitaremos utilizar un comando del programa exonerate, fastatranslate. Sin embargo, el archivo generado contiene todos los marcos de lectura posibles (tres fordward y tres reverse) de los que tendremos que seleccionar el que corresponda a nuestra proteína. Esto lo hemos automatizado pero puede realizarse manualmente introduciendo por ejemplo -F 1, si queremos el primer marco de lectura.
![]()
Genewise
GeneWise es una herramienta que también se utiliza para predicción de genes, como alternativa a exonerate. Permite comparar una secuencia de proteína (query) contra una secuencia de DNA genómico.
Para llamar al programa utilizamos el siguiente comando:
Genewise admite diversos comandos, estos son los que hemos utilizado:
- -pep: muestra la traducción a proteína.
- -pretty: muestra el alineamiento pretty ASCII.
- -cdna: muestra la secuencia cDNA predicha.
- -gff: muestra la predicción del gen en formato GFF.
Hay más informacion sobre genewise y sus parámetros en la página principal.
Alineación múltiple de secuencias
T-coffee
Tras obtener nuestras secuencias proteicas candidatas debemos realizar un alineamiento de cada una con la query inicial para determinar si hay cambios en la secuencia de nucleótidos. Para este propósito empleamos el programa t-coffee, en el que podremos ver el grado de homología entre nuestras dos proteínas. Este programa lo ejecutamos con el siguiente comando:
![]()
Predicción de elementos SECIS y Seblastian
Las selenoproteínas eucariotas contienen unas estructuras con forma de loop denominadas SECIS (Sec insertion sequence). Secuencia necesaria para la incorporación de la selenocisteina (U) durante la traducción, evitando que el ribosoma interprete el codón UGA como un STOP.
Para la caracterización de las selenoproteínas encontradas hemos realizado una búsqueda de elementos SECIS mediante el uso de la herramienta SECISearch3. Este es un método de predicción basado en las propiedades de la estructura primaria y secundaria que los elementos SECIS presentan. SECISearch3 tiene una gran sensibilidad, sin embargo, hay que tener en cuenta que su especificidad es baja y son muchos los hits que se encuentran, por lo que los datos deben ser analizados manualmente.
Seblastian es un programa online de predicción de genes de selenoproteínas que utiliza la herramienta SECISearch3, para buscar los genes en la región upstream del elemento SECIS [8]. En nuestro trabajo hemos utilizado esta herramienta para validar nuestros resultados.
Podemos encontrar ambos programas en la siguiente dirección web.
Scripts y automatización
Programa
El siguiente programa se ha utilizado para automatizar todos los procesos anteriormente descritos:

Bash
Este archivo bash contiene los comandos necesarios para crear los paths para usar los programas (descritos al inicio) y para llamar al programa de automatización.
![]()