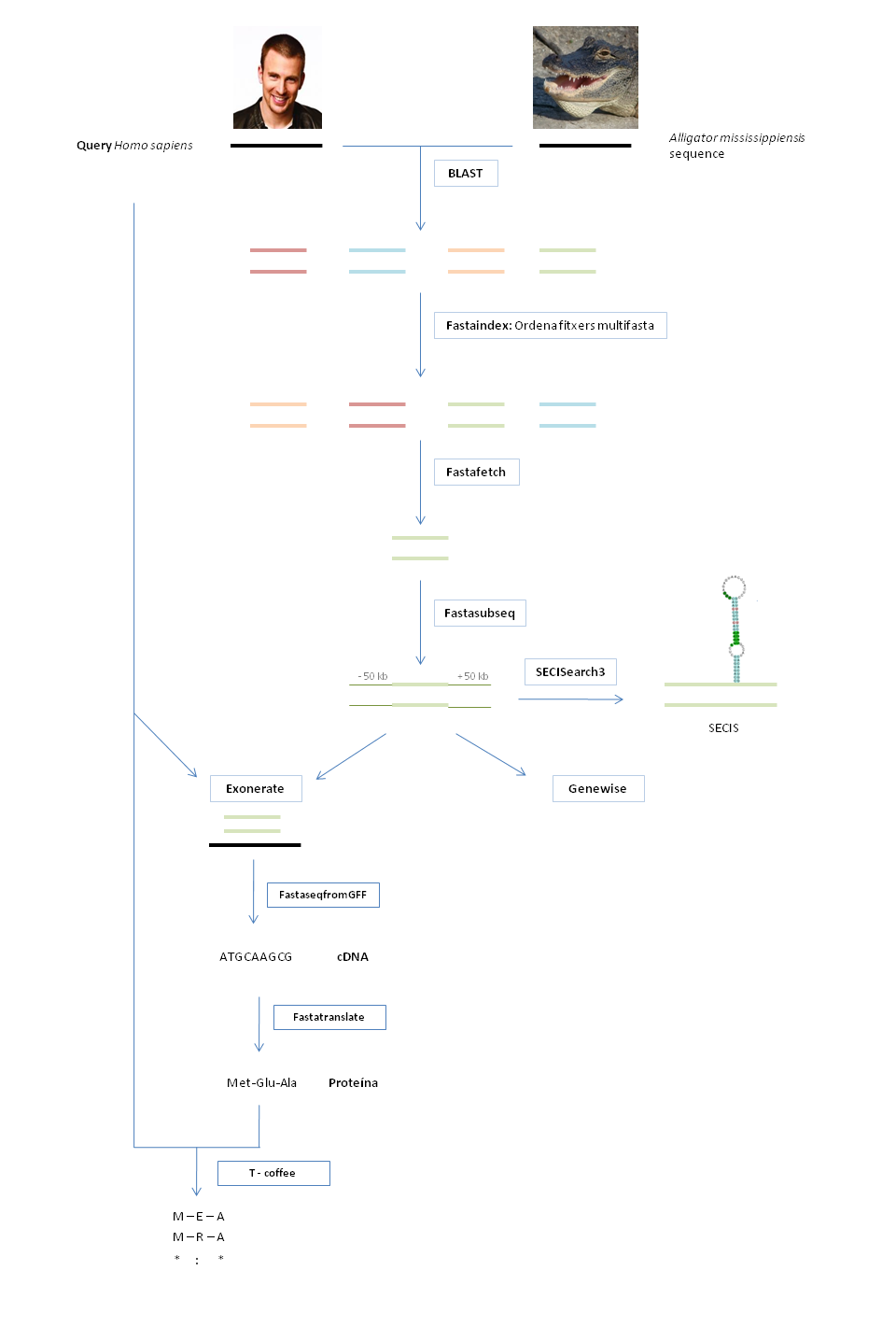
El objetivo de nuestro proyecto es realizar una búsqueda de selenoproteínas en el genoma de Alligator mississippiensis. A continuación, se detallan los materiales y métodos que se han llevado a cabo para el desarrollo del proyecto y se especifican los comandos básicos utilizados.
En nuestro caso, el genoma de Alligator mississippiensis fue proporcionado por el profesorado de la asignatura en el siguiente directorio:
Se trata de un fichero de gran tamaño y complejo de manipular, motivo por el cual se nos facilitó esta información. Sin embargo, se encuentra disponible en la base de datos Ensembl.
Con el objetivo de obtener las secuencias de las selenoproteínas en Homo sapiens, se ha recurrido a la base de datos SelenoDB2.0. Las secuencias de las selenoproteínas humanas se han utilizado como querys, que se han alineado con el genoma de Alligator mississippiensis para saber si estas secuencias se encuentran también en la especie de interés. Sin embargo, cabe tener en cuenta que Alligator mississippiensis es una especie filogenéticamente alejada de Homo sapiens. Para las seleproteínas SelK, SelN y SelP, se han buscado querys de la especie Pelodiscus sinensis, que es más cercana a Alligator mississippiensis.
Las querys de las selenoproteínas contienen una selenocisteína en su secuencia, que se representa con la letra U (codón TGA). Es necesario cambiar la U por una X en cada query para que los programas funcionen correctamente.
BLAST (Basic Local Alignment Search Tool) es un programa informático que permite realizar alineamientos locales de una secuencia problema con un conjunto de secuencias utilizadas como referencia extraídas de una base de datos. El programa utiliza un algoritmo heurístico capaz de seleccionar las secuencias o hits con mayor similitud a la query.
Existen distintos tipos de BLAST según la naturaleza de las secuencias que se pretende comparar. En nuestro caso, se ha utilizado tBLASTn, que permite alinear las secuencias proteicas (querys) de las especies tomadas como referencia con una base de datos de secuencias nucleotídicas, que en este caso es el genoma de Alligator mississippiensis.
Para acceder al tBLASTn, primeramente es necesario introducir los permisos de exportación del programa, que se muestran a continuación:
Para ejecutar el programa, se utilizó la siguiente comanda:
-p es el tipo de BLAST utilizado, -i es la localización de la query, -d es el genoma del Alligator mississippiensis, y -o es el output o fichero donde se almacena el resultado final (output). El output consiste en un listado de los hits que tienen mayor similitud con la query con la que se ha alineado el genoma. La significancia estadística de cada hit se expresa a través del valor E (E-value), que es la probabilidad de encontrar un cierto hit mediante BLAST únicamente debido al azar. Un alineamiento será significativo siempre que el E-value sea igual o inferior a 10-4.
Una vez se han obtenido los resultados del tBLASTn (hits), se ha considerado el hit con mejor E-value, se han tomado las posiciones nucleotídicas inicial y final de ese hit y se han expandido 50 Kb upstream y 50 Kb downstream. Esto nos permite delimitar una región en la que se asegura que esté el gen de interés. La extracción de estas secuencias más cortas implica tener el genoma indexado, con el siguiente comando:
Fastaindex es el programa que indexa el genoma que se encuentra en el directorio donde se encuentran los cromosomas del genoma de Alligator mississippiensis que se indica (directori/genome.fa), y sortida.index es la ubicación del fichero de salida.
Seguidamente, se ejecutó el programa Fastafetch, que permite la extracción del scaffold donde se encuentran cada uno de los hits estadísticamente significativos obtenidos mediante el tBLASTn.
Finalmente, se ha ejecutado el programa Fastasubseq, que permite seleccionar la región que rodea el gen de interés.
Siendo start el nucleótido por el que empieza la subsecuencia (50kb upstrean del inicio predicho por tBLASTn), length, la longitud de nucleótidos que incluye la subsecuencia (100.000) y genomic.fa la ubicación del fichero de salida.
Se han utilizada dos programas de predicción de genes, Exonerate y Genewise, que utilizan algoritmos distintos, por lo que los resultados obtenidos varían entre uno y otro.
ExonerateCon el objetivo de predecir los genes de interés en el genoma de Alligator mississippiensis, se ha utilizado el software Exonerate. Primeramente, se ha introducido el permiso de exportación:
A continuación, se ejecutó el siguiente comando:
Siendo -m p2g el modelo de alineamiento, --showtargetgff el comando que incluye el resultado en formato GFF en el fichero de salida, -q, la ubicación de la query, -t, la subsecuencia y sortida.gff, la ubicación del fichero en formato GFF. Además, como queremos que el fichero de salida solo incluya los exones, se ha añadido el comando: egrep, que selecciona las líneas donde aparece el patrón definido por el argumento -w (exon, en nuestro caso), y sortidadm.gff, que es el fichero de salida. Se utilizó el programa FastaseqfromGFF.pl para extraer los resultados en formato FASTA, ya que construye una secuencia de nucleótidos a partir de un fichero GFF.
Siendo genomic.fa la subsecuencia, sortidadm.gff el fichero que contiene los exones, y sortidadm.cdna el fichero que contiene la secuencia de cDNA en formato FASTA.
Finalmente, se ejecutó el programa Fastatranslate para obtener la secuencia de aminoácidos a partir del cDNA.
Siendo sortidadm.cdna la ubicación de la secuencia de nucleótidos de cDNA, y translate.fa el fichero de salida en formato multifasta. El argumento -F 1 limita el marco de lectura elegido, que en este caso, es el primero de los que ofrece el programa FastaseqfromGFF.
GenewiseDespués de la obtención de las predicciones con el software exonerate, se ha ejecutado el software Genewise con los siguientes permisos de exportación y comando de ejecución:
siendo -pep la función utilizada para que en el fichero sortida.gff se muestre la secuencia peptídica predicha; -pretty la función para que muestre el alineamiento; -cdna la función para que muestre la secuencia genómica alineada; -gff la función para que la información se muestre en formato gff; -both la función para que la predicción se lleve a cabo en los dos sentidos de lectura (forward i reverse); query.fa la ubicación de la query; genomic.fa la subsecuencia extraída anteriormente y sortida.gff el fichero de salida.
T-CoffeeSe ha utilitzado el software T-Coffee (Tree-based Consistency Objective Function for alignment Evaluation). Este programa permite realizar un alineamiento global de la proteína de interés obtenida del genoma de Alligator mississippiensis con la secuencia query utilizada en cada caso. Este alineamiento permite identificar posición a posición con que aminoácido se alinea la proteína en la especie de interés. Con ello, se podrá concluir si se trata de selenoproteínas o proteínas homólogas en cisteína. El software T-Coffee se ha ejecutado con el siguiente comando:
siendo align.blast la query, translate.fa, la secuencia de aminoácidos obtenida, y align.fa, el fichero de salida, que contiene el alineamiento entre la query y la proteína en la especie de interés.
Después de la identificación de las proteínas en el genoma de Alligator mississippiensis, se ha realizado una búsqueda de elementos SECIS (Selenocysteine Insertion Sequence), que, junto con el codón UGA, es el otro criterio utilizado para la caracterización de selenoproteínas, ya que mejora la credibilidad de las predicciones realizadas anteriormente. La búsqueda de elementos SECIS se ha llevado a cabo a través del software SECISearch3 online, introduciendo el archivo Fastasubseq. Se han tomado como válidos los elementos SECIS que se encuentran dentro de los límites de unos pocos pares de bases y 5-10 Kb como máximo de distancia, respecto a la selenocisteína. Se eligieron los elementos SECIS con mejor score, siendo este preferiblemente superior a 15.