| |
|
|
| |
|
|
| |
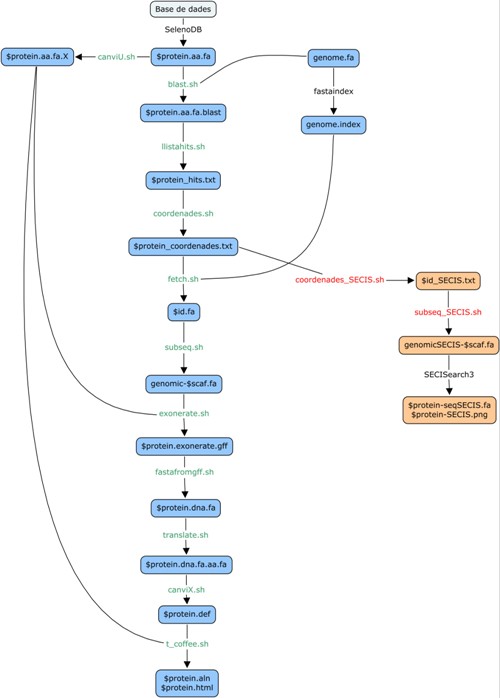
L’objectiu del nostre treball consisteix en identificar totes les selenoproteïnes de l’Orycteropus afer. El procediment que s’ha seguit es divideix en diferents apartats, que alhora coincideixen amb els diferents programes realitzats i utilitzats.
Per a la realització de tot el procediment s’ha treballat a partir del clúster de docència de la UPF, format per dos ordinadors amb un total de 16 processadors engegats les 24 hores del dia. Per tal d’entrar-hi s’ha de fer mitjançant la següent ordre:
$ ssh -x UXXXXX@luke.upf.edu
S’han realitzat 11 programes bash:
I 5 programes perl:
Per a descarregar-te tots els programes en format .zip, fes clic aquí.
Ara bé, com que el cúster de docència i els ordinadors que hem utilitzat estan basats en arquitectures diferents, per fer anar els mateixs programes s’hauran d’executar aquestes comandes:
$ export PATH=/cursos/BI/bin/ncbiblast/bin:$PATH# pel NCBI Blast (executar abans del blast.sh)
$ cp /cursos/BI/bin/ncbiblast/.ncbirc ~/# pel NCBI Blast (executar abans del blast.sh)
$ export PATH=/cursos/BI/soft/exonerate/i386/bin:$PATH# per l'exonerate (executar abans del exonerate.sh)
$ export PATH=/cursos/BI/bin:$PATH# pel GeneWise, el fastaseqfromGFF.pl i el t_coffee (executar abans del fastafromgff.sh i el t_coffee.sh)
Els passos que s’han seguit son:
|
|
| |
Obtenció de les selenoproteïnes (querys)a
La query o seqüència model és la proteïna o regió proteica coneguda que intentarem relacionar amb els genomes en estudi. En el nostre cas, les querys seran les selenoproteïnes ja anotades d’altres organismes.
Inicialment es va escollir com a query les selenoproteïnes de l’humà, i ens vam descarregar les seqüències d’aquestes, ja que actualment són les que estan millor anotades. A més, és el mamífer amb més selenoproteïnes anotades en la base SelenoDB.
Posteriorment, vam ampliar el rang de seqüències utilitzades. Vam utilitzar algunes de les selenoproteïnes descrites i anotades a T. nigroviridis, ja que entre aquestes n’hi trobem algunes (SelU, SelL, Fep15) que els humans no tenim anotades.
Finalment, també vam baixar-nos i analitzar les selenoproteïnes de Loxodonta (una espècie d’elefant) perquè està més proper evolutivament a Orycteropus afer que els humans
.Per a obtenir aquestes seqüències hem anat a SelenoDB |
Obtenció del genoma de l’Orycteropus afera
El genoma d’Orycteropus afer utilitzat es troba al directori:
/cursos/BI/genomes/project_2014/Orycteropus_afer
També el podríem obtenir des de l ’Ensembl |
|
Blast
El BLAST (Basic Local Alignment Search Tool) és un algorisme que realitza alineaments locals entre una query (ja sigui de DNA o proteica) i un conjunt de seqüències determinades, que podem trobar, per exemple, en una base de dades. Aquest és un algorisme heurístic, i basa la seva cerca en aquells alineaments amb alt grau de similaritat. Així doncs, pot ser que no puguem detectar els hits reals que presentin una similaritat menor del llindar que hem establert com a fiable.
En funció de si les seqüències a comparar són proteiques o genòmiques s'utilitza un flavour determinat de BLAST. En el nostre cas, com que volem alinear querys proteiques contra un genoma, utilitzem el tBLASTn.
Com s’ha comentat a l’inici, per executar aquest programa s’ha d’extreure el software necessari amb les següents ordres al shell: |
$ export PATH=/cursos/BI/bin/ncbiblast/bin:$PATH
$ cp /cursos/BI/bin/ncbiblast/.ncbirc ~/
Per tal d'automatitzar el procés s'ha fet un programa bash (blast.sh): |
#!/bin/bash
# al principi per triar la proteïna analitzada utilitzavem això:
# echo quina proteina vols analitzar?
# read PROTEINA
# però vam veure que era més ràpid i eficaç d’aquesta altra manera:
PROTEINA=$1
USER=$2
for protein in $PROTEINA/*.fa; do
echo $protein
blastall -m 8 -p tblastn -i /homes/users/$USER/selenoproteins/$protein -d /cursos/BI/genomes/project_2014/Orycteropus_afer/genome.fa -o $protein.blast
done
On:
-m ens indica el format de l’output (en aquest cas –m 8, format columnar, indica que ens sortirà indexat en forma de taula però sense el camp de la primera fila que correspondria a l’explicació del que és cada columna).
-p ens indica la modalitat de blast utilitzada
-i (input) ens indica quina és la query
-d (database) ens indica quina és la base de dades
-o (output) dóna nom al fitxer de sortida del BLAST
Per tant, per executar-lo s’haurà d’escriure al shell la ordre:
$ ./blast.sh PROTEINA (ex:DI1) USER (ex:U74747)
Un cop passat aquest programa tindrem un fitxer, anomenat $protein.aa.fa.blast, on hi apareixeran tots els hits que s’hagin trobat en els alineaments de cada selenoproteïna. La taula obtinguda doncs, mostrarà diferents camps referent a aquests hits: la identitat de la query, regió on es troba el hit, percentatge d’identitat, longitud d’alineament, mismatches, gaps openings, inici de l’alineament dins la query, final de l’alineament dins la query, inici dins el genoma, final dins el genoma, e-value, i bit-score (puntuació) de l’alineament.
Cada BLAST serà guardat dins la carpeta de la seva query.
|
Fasta índex |
Per extreure la regió genòmica d’interès realitzarem diferents passos: Fastaindex, selecció de hits i coordenades, Fastafetch i Fastasubseq.
El Fastaindex ordena el genoma en diferents regions i fa un índex de les seqüències en fasta. En el nostre cas ordena el genoma per scaffolds.
Com que el genoma de l’Orycteropus afer ja estava indexat no ha calgut realitzar cap programa, simplement localitzar el fitxer orycteropus_afer.index al luke.
|
Selecció hits |
En aquest pas vam seleccionar els hits significatius. Per això del resultat del BLAST ens quedem només amb aquells hits que tinguin un e-value inferior a e-10, i en cas que no n’hi hagués, qualsevol que estigui entre e-10 i e-5. Per tal d’optimitzar el procés fem el programa Perl (selecciohits.pl): |
#!/usr/bin/perl -w
use strict;
my @hit;
my $comptador;
$comptador = 0;
while (<STDIN>) {
@hit = split("\t");
if ($hit[10] <= 1e-10){
print $_;
$comptador = $comptador + 1;
}
elsif ($hit[10] > 1e-10 && $hit[10] < 1e-5 && $comptador == 0){
print $_;
}
|
I un programa bash perquè ens vagi executant el programa anterior en les nostres proteïnes (llistahits.sh): |
#!/bin/bash
PROTEINA=$1
for protein in $PROTEINA/*.fa.blast; do
echo $protein
./selecciohits.pl < $protein > $protein"_hits.txt"
done
|
Així executarem al shell:
$ ./llistahits.sh PROTEINA (ex.DI1)
i n’obtindrem un output, en la carpeta de la proteïna en qüestió, anomenat $protein_hits.txt.
|
Selecció de coordenades
|
Aquest pas ens permet trobar a quines coordenades del genoma correspon la nostra query, i per tant seleccionar l’inici i el final en el genoma de l’Orycteropus afer del que correspondria a cada hit. En cas que el final fos un nombre menor que l’inici, significarà que el hit es troba a la cadena reversa i per tant cal intercanviar els valors de les variables “inici” i “final”. Per això realitzem un programa perl (coordenades.pl):
|
|
|
|
| |
my @row=();
my $ini=0;
my $fin=0;
my %hash_ini = ();
my %hash_fin = ();
while (<STDIN>)
{
chomp($_);
@row=split(/\t/,$_);
if ($row[8] < $row[9])
{
$ini=$row[8];
$fin=$row[9];
}
else
{
$ini=$row[9];
$fin=$row[8];
}
#%ini= ($row[1], $ini);
#%fin= ($row[1], $fin);
if (exists $hash_ini{$row[1]}) {
if ($ini < $hash_ini{$row[1]}){
$hash_ini{$row[1]} = $ini;
}
}
else {
$hash_ini{$row[1]} = $ini;
}
if (exists $hash_fin{$row[1]}) {
if ($fin > $hash_fin{$row[1]}){
$hash_fin{$row[1]} = $fin;
}
}
else {
$hash_fin{$row[1]} = $fin;
}
}
|
|
| |
I un programa bash per anar executant el perl (coordenades.sh):
|
|
| |
#!/bin/bash
PROTEINA=$1
for protein in $PROTEINA/*.txt;do
echo $protein
./coordenades.pl < $protein > $protein"_coordenades.txt"
done
|
|
| |
Per executar-lo posarem al shell:
./coordenades.sh PROTEINA (ex.DI1)
I n’obtindrem un output anomenat $protein_coordenades.txt. |
|
| |
|
|
| |
Fasta Fetch
|
|
| |
Per tal de passar les coordenades a través del genoma i poder generar arxius que continguin les regions genòmiques d’interès, hem realitzat el seguent programa bash (fetch.sh):
|
|
| |
#!/bin/bash
PROTEINA=$1
FILE=$2
for protein in $PROTEINA/*coordenades.txt; do
# echo $protein
for id in $(cut -f1 $protein); do
fastafetch /cursos/BI/genomes/project_2014/Orycteropus_afer/genome.fa $FILE/orycteropus_afer.index $id > $PROTEINA/$id.fa
echo $protein:$id
done
done
|
|
| |
On:
- genome.fa és el genoma de l’organisme
- orycteropus_afer.index és el genoma indexat
- $id el hit d’interès, indicant el nom de l’scaffold on es troba
S’executarà escrivint la següent ordre al shell:
./fetch.sh PROTEINA (ex.DI1) FILE (ex.U74747)
i n’obtindrem un output anomenat $id.fa que ens indicarà en quin scaffold està la seqüència d’interés.
|
|
| |
Fasta Subseq |
|
| |
Finalment podem obtenir la regió d’interés gràcies al Fastasubseq, que encara delimita més aquesta regió obtenint-ne seqüències més curtes. El programa realitzat és un bash (subseq.sh): |
|
| |
#!/bin/bash
PROTEINA=$1
for protein in $PROTEINA/*coordenades.txt; do
for scaf in $(cut -f1 $protein); do
start=$(grep $scaf $protein | cut -f2);
start=$(expr $start - 100000);
if [ $start -lt 0 ]; then
start=1;
fi;
end=$(grep $scaf $protein | cut -f3);
end=$(expr $end + 100000);
length=$(expr $end - $start);
fastasubseq $PROTEINA/$scaf.fa $start $length > $PROTEINA/genomic-$scaf.fa
echo $scaf
done
done
|
|
| |
On:
Els paràmetres start i lengh s’extreuen a partir dels fitxers que contenen les coordenades d’interès (generats a partir de l’output del BLAST). S’ha mirat on comença i acaba l’aliniament dine el genoma de l’O.afer de cada hit significatiu. Per assegurar-nos que la seubseqüència inclou el gen d’interès sencer hem expendit els marges per davant i darrera de l’alineament, considerant la posició inicial de la subseqüència 100.000* nucleòtids upstream de la posició inicial i la posició final 100.000* nucleòtids downstream de la posició final de l’alineament.
Per tal d’executar-lo cal la ordre al shell:
$ ./subseq.sh PROTEINA (Ex.DI1)
I aquesta seqüència (output) la desa en un fitxer anomenat genòmic-$scaf.fa
*Al principi es va realitzar amb 10.000 nucleòtids per banda, però per a millorar els resultats i agafar talls que no s’alineaven al principi o al final vam augmentar aquest nombre.
|
|
| |
Canviar les U per les X |
|
| |
S’han de canviar les U de la nostra query per X, doncs l’Exonerate no reconeix el símbol U (selenocisteina). Per tal de realitzar aquest canvi es fa un programa perl (canviU.pl):
|
|
| |
#! /usr/bin/perl -w
my @seq=();
my $aa="";
while (<STDIN>) {
chomp($_);
if ($_!~m/>/){
@seq=split(//,$_);
foreach $aa (@seq){
if ($aa eq "U"){
print "X";
}elsif(($aa eq "#")||($aa eq "@")||($aa eq "%")||($aa eq " ")){
next;
}else{
print"$aa";
}
}
print "\n";
}else{
print "$_\n";
}
}
|
|
| |
I per executar-lo de manera automàtica, un programa bash (canviU.sh): |
|
| |
#!/bin/bash
PROTEINA=$1
for protein in $PROTEINA/*.aa.fa; do
echo $protein
./canviU.pl < $protein > $protein.X
done
|
|
| |
S’executarà posant l’ordre al shell:
$ ./canviU.sh PROTEINA (ex.DI1)
|
|
| |
Exonerate |
|
| |
Un cop ja tenim les regions genòmiques d’interès en fitxers executem el programa Exonerate. Aquest compara seqüències de manera general i ràpida, però també ens permet alinear seqüències utilitzant la programació dinàmica exhaustiva, necessària en alguns casos. Si no, per defecte utilitza els models heurístics. A més, prediu l’estructura exònica de la seqüència introduïda a partir de l’alineament amb la query.
Així doncs compararem la nostra query amb la regió genòmica on hi ha la possible selenoproteïna gràcies a un programa bash (exonerate.sh):
|
|
| |
#!/bin/bash
PROTEINA=$1
for protein in $PROTEINA/*.aa.fa.X; do
# perquè ens ho faci només amb la regió genòmica que volguem i no amb totes (e-value més gran):
echo quin genomic vols?
read genomic
exonerate -m p2g --showtargetgff -q $protein -t $genomic | egrep -w exon > $protein.exonerate.gff
echo $genomic
done
echo $protein
done
|
|
| |
On:
-m fa referència al model de l’alineament a utilizar (p2g significa protein to genome, per tant comparem una seqüència proteica amb una seqüència de DNA).
-q fa referència a la query utilitzada
-t fa referència a la seqüència comparada
la comanda després del pipe (|) fa referència a ala selecció de les seqüències exòniques d’interès per després passar-les a cDNA.
S’haurà d’executar amb la següent ordre al shell:
$ ./exonerate.sh PROTEINA (ex.DI1)
i l’output serà un fitxer anomenat $protein.exonerate.gff
|
|
| |
FastaseqfromGFF |
|
| |
Un cop tenim les seqüències exòniques en format GFF, hem utilitzat el programa FastaseqfromGFF.pl per tal d’obtenir el cDNA en format fasta. Com hem dit abans s’ha d’inicialitzar el programa amb l’ordre següent al shell:
$ export PATH=/cursos/BI/bin:$PATH
Per tal d’automatitzar-lo fem un programa bash (fastafromgff.sh):
|
|
| |
#!/bin/bash
PROTEINA=$1
for file in $PROTEINA/*.exonerate.gff; do
for genomic in $PROTEINA/genomic*.fa; do
fastaseqfromGFF.pl $genomic $file > $file.dna.fa
echo $genomic
done
|
|
| |
Després d’executar-lo mitjançant l’ordre:
$ ./fastafromgff.sh PROTEINA (ex.DI1)
obtenim un fitxer $proteina.dna.fa que contindrà el cDNA d’interès.
|
|
| |
Translate |
|
| |
En aquest pas traduïm la regió exònica a proteïna gràcies al Fastatranslate. D’aquesta manera s’obté una seqüència d’aminoàcids amb els sis marcs de lectura (ORFs) possibles. Després d’analitzar els diferents ORFs hem seleccionat aquell que ens interessa, és a dir aquell que només tingui un asterisc (el de la selenocisteïna, si en tenen més significa que corresponen a codons stop). Com que hem vist que coincidia quasi sempre amb el transcrit 1, ja hem aplicat això a l’ordre i agafem directament aquest primer. Ara bé, es comprova i s’analitza després perquè no es cometin errors.
El programa a executar és el següent (translate.sh):
|
|
| |
#!/bin/bash
PROTEINA=$1
for mRNA in $PROTEINA/*.dna.fa; do
fastatranslate -F 1 $mRNA > $mRNA.aa.fa
echo $mRNA
done
|
|
| |
On:
-F fa referència a seqüència forward (1 és el nombre de pauta a seguir)
Després d’executar aquest programa de la següent manera:
$ ./translate.sh PROTEINA(ex. DI1)
S’obté un arxiu anomenat $protein.dna.fa.aa.fa, que contindrà la seqüència proteica.
|
|
| |
Canvi de U per X |
|
| |
Per tal de poder alinear correctament i veure’n el resultat de manera correcta, canviem les U d’aquesta subseqüència que hem generat a X. Si no s’executés a l’hora de traduir es canviarien les U per * de manera que al alinear ens sortiria una X (de la query) aliniada amb un –, resultat que es pot confondre ja que aquest símbol ens indica un gap i no una selenocisteïna com nosaltres volem.
El programa perl (canviX.pl):
|
|
| |
#! /usr/bin/perl -w
my @seq=();
my $aa="";
while (<STDIN>) {
chomp($_);
if ($_!~m/>/){
@seq=split(//,$_);
foreach $aa (@seq){
if ($aa eq "*"){
print "X";
}elsif(($aa eq "#")||($aa eq "@")||($aa eq "%")||($aa eq " ")){
next;
}else{
print"$aa";
}
}
print "\n";
}else{
print "$_\n";
}
}
|
|
| |
I
el programa bash (canviX.sh): |
|
| |
#!/bin/bash
PROTEINA=$1
for protein in $PROTEINA/*.dna.fa.aa.fa; do
echo $protein
output=$(echo $protein | cut -d "." -f1)
./canviX.pl < $protein > $output.def
done
|
|
| |
T-Coffe |
|
| |
El programa T-Coffee (Tree-based Consistency Objective Function for alignment Evaluation) permet realitzar alineaments globals múltiples utilitzant un mètode progressiu que aparella seqüències similars i usa el resultat com a punt de partida del següent aparellament. Nosaltres l'hem fet servir per alinear la nostra query amb les selenoproteïnes o els homòlegs de cisteïna obtinguts a partir de l'Exonerate un cop ja han sigut traduïts, per tant, comparem dues seqüències proteiques.
El programa bash que hem generat per a l’automatització d’aquest és (t_coffee.sh): |
|
| |
#!/bin/bash
PROTEINA=$1
for seq1 in $PROTEINA/*.def; do
seq2=$(echo $seq1 | cut -d "." -f 1).aa.fa.X
t_coffee $seq1 $seq2
echo $seq1
echo $seq2
done
|
|
| |
Executant-lo amb aquesta ordre al shell:
$ ./t_coffee.sh PROTEINA
Obtenim un fitxer de sortida en format $protein.html i un altre en $protein.aln. En aquests fitxers hi podrem veure l’alineament en qüestió, així doncs, ens permetrà analitzar-ne la similaritat entre la query i la proteïna de l’O.afer. El fet de mostrar una puntuació de l’alineament (score) fa que ens doni una idea de la fiabilitat del resultat.
Si en aquest alineament veiem una semblança significativa entre les dues proteïnes i veiem que hi ha selenocisteïnes alineades (X amb X) entre elles podem tenir indicis que ens trobem davant una selenoproteïna. Ara bé, es realitzen altres passos de complementació per assegurar-nos-en del tot: buscar elements SECIS i maquinària de transcripció necessària per a la síntesi d’aquestes, modificar paràmetres per obtenir informació més fiable, etc.
|
|
| |
Complementació |
|
| |
Millora dels resultats obtinguts |
|
| |
Després d'analitzar els primers programes obtinguts, s'han modificat alguns paràmetres dels programes per tal de millorar aquelles proteïnes de les quals s'ha obtingut un resultat dubtós. Un dels procediments que s'ha seguit és augmentar la regió genòmica que agafem amb el programa “fastasubseq.sh” per tal d'assegurar-nos que trobem el gen de la proteïna en la regió estudiada.
Per altra banda, en alguns casos també s'ha utilitzat la opció –exhaustive de l'exonerate; que considera més alineaments, tot i no ser tan significatius, i per tant ens permet trobar alineaments que en un principi no hauríem tingut en compte.
|
|
| |
Utilització de querys d'una espècie més propera filogenèticament
|
|
| |
Tot i escollir el H.sapiens com a espècie de referència per a obtenir les querys, ja que és una de les espècies amb el genoma més ben anotat, hem cregut oportú realitzar també l'anàlisi amb una altra espècie més propera filogenèticament. Després de consultar-ho a l'apartat de Taxonomia del NCBI , s'ha escollit el Loxodonta africana com l'espècie del SelenoDB més propera a Orycteropus afer.
|
|
| |
Cerca d'homòlegs en cisteïna |
|
| |
Sabem que les selenoproteïnes contenen un àtom de seleni enlloc de sofre, en les cisteïnes de la seva seqüència; i per tant podem trobar proteïnes homòlogues amb cisteïna enlloc de selenocisteïna. Així, una bona aproximació per a trobar selenoproteïnes en el genoma de O.afer és realitzar el mateix procés amb homòlegs amb cisteïna dels organismes escollits (Homo sapiens i Loxodonta africana), ja que pot ser que en l'organisme estudiat (O.afer) hi trobem una selenocisteïna enlloc d'una cisteïna.
|
|
| |
Cerca d'elements SECIS |
|
| |
Els elements SECIS són estructures essencials per a la síntesi de selenoproteïnes, i per tant si en podem identificar en la regió 3'-UTR dels gens que les codifiquen, podrem estar més segurs que la predicció és correcta.
Com que sabem que aquests elements es troben a la regió 3'-UTR, s'han generat noves seqüències genòmiques d'aquesta regió.
Primer de tot s'han establert les coordenades de la regió genòmica a analitzar, essent l'inici el valor de la variable “final” de les coordenades de la regió genòmica on es localitza el gen, i el final, 10 kilobases més enllà. Aquest procés s'ha automatitzat amb un programa Perl:
|
|
| |
#! /usr/bin/perl -w
my @row=();
my $ini=0;
my $fin=0;
my $id;
while (<STDIN>)
{
chomp($_);
@row=split(/\t/,$_);
$id= $row[0];
$ini= $row[2];
$fin=$ini+10000;
print $id,"\t", $ini,"\t",$fin,"\n";
}
|
|
| |
Aquest programa s'ha passat per a totes les proteïnes gràcies a un programa bash: |
|
| |
#!/bin/bash
PROTEINA=$1
for coordenades in $PROTEINA/*coordenades.txt; do
./coordenades_SECIS.pl < $coordenades > "$coordenades"_SECIS.txt
echo $PROTEINA:$coordenades
done
|
|
| |
Llavors s'ha utilitzat el fastasubseq per a obtenir la regió genòmica desitjada a partir de les coordenades anteriors. El procés s'ha automatitzat amb un programa bash: |
|
| |
#!/bin/bash
PROTEINA=$1
for protein in $PROTEINA/*SECIS.txt; do
for scaf in $(cut -f1 $protein); do
start=$(grep $scaf $protein | cut -f2);
end=$(grep $scaf $protein | cut -f3);
length=$(expr $end - $start);
fastasubseq $PROTEINA/$scaf.fa $start $length > $PROTEINA/genomicSECIS-$scaf.fa
echo $scaf
done
done
|
|
| |
Finalment, s'han analitzat les regions genòmiques obtingudes a partir del programa SECISearch3, que prediu la seqüència i estructura dels elements SECIS a la regió. |
|
| |
Predicció de selenoproteïnes mitjançant SEBLASTIAN |
|
| |
Seblastian és un algorisme dissenyat per investigadors del CRG (referència article) que prediu la seqüència de selenoproteïnes en una regió genòmica determinada, a partir de la predicció d'elements SECIS en aquesta regió i la similaritat amb selenoproteïnes conegudes de diferents espècies.
Hem utilitzat aquest recurs per a corroborar les nostres prediccions i millorar els resultats de les selenoproteïnes de les quals havíem obtingut resultats dubtosos.
En aquest diagrama es pot visualitzar un esquema de tot el procés explicat anteriorment: |
|
| |
 |
|
| |
|
|
