
Com ja hem dit, l'objectiu del nostre estudi era trobar en el genoma de Myotis davidii les selenoproteïnes, així com la maquinària d'aquest. Per fer-ho, vam comparar les selenoproteïnes ja anotades d'altres espècies amb el nostre genoma (Homo sapiens i Myotis lucifugus, mitjançant les comandes per a shell que s'expliquen a continuació.
D'altra banda, per tal d'agilitzar la nostra recerca vam elaborar un programa en llenguatge perl que feia la cerca donada una query. Podeu consultar-lo aquí.
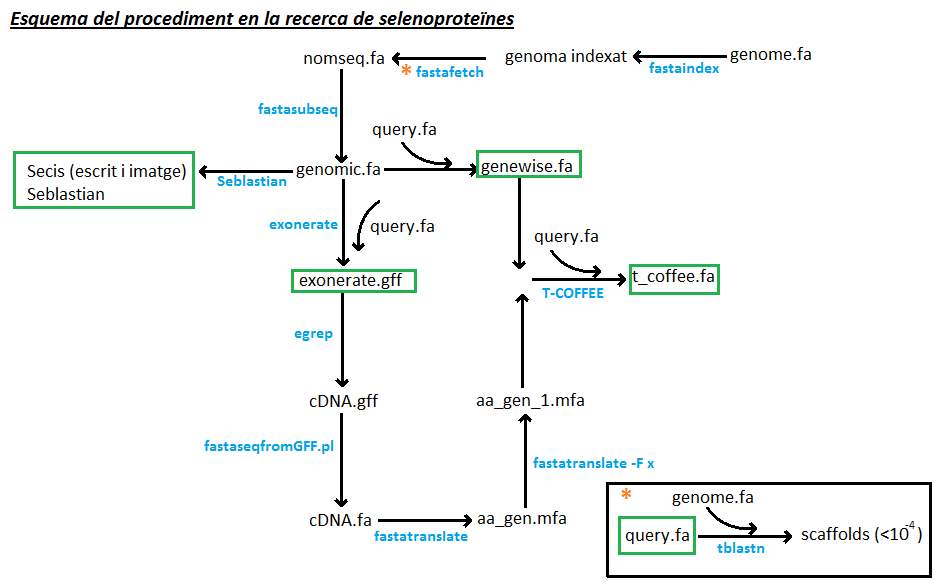
Esquema 1. En aquest esquema es mostra els passos que s'han dut a terme per anotar les selenoproteïnes. Els marcs de color verd, assenyalen els arxius que podem trobar en l'apartat de resultats.
1. Obtenció de les queries:
Per determinar quines selenoproteïnes (i proteïnes de maquinària) són presents al genoma de Myotis davidii, en primer lloc hem escollit la query adequada per comparar-la amb el nostre genoma d'estudi. Aquestes queries les hem obtingut de la base de dades SelenoDB, concretament del genoma de Myotis lucifugus, ja que aquesta espècie pertany al mateix gènere que la nostra, i per tant, hauria de tenir selenoproteïnes similiars. Tot i així, no totes les selenoproteïnes estaven ben anotades i per tant, vam recórrer a cercar-les a la base de dades del NBCI. En el cas de no trobar-les al NBCI vam utilitzar les queries de les selenoproteïnes de l'Homo sapiens, l'animal més ben anotat.
Cal remarcar, que hem hagut de modificar aquelles queries que continguessin una selenocisteïna (U), ja que el programa utilitzat no reconeixia el seu caràcter. Per tant, hem substituït el caràcter U pel caràcter X.
Podeu trobar totes les queries usades aquí.
2. Obtenció del genoma de Myotis davidii:
Els professors de l'assignatura ens van facilitar el genoma de Myotis davidii, utilitzat per elaborar aquest treball, extret mitjançant la següent comanda en el terminal/shell:
| $ formatdb –i cursos/BI/genomes/project_2014/Myotis_davidii/genome.fa |
3. Cerca manual de les selenoproteïnes:
Amb la finalitat de realitzar la cerca manual de selenoproteïnes hem hagut d'exportar diversos programes recollits en la següent taula, juntament amb la comanda utilitzada:
| Programes exportats | Comanda d'exportació |
| NCBI Blast | $ export PATH=/cursos/BI/bin/ncbiblast/bin:$PATH |
| NCBI Blast | $ cp /cursos/BI/bin/ncbiblast/.ncbirc ~/ |
| Exonerate | $ export PATH=/cursos/BI/soft/exonerate/i386/bin:$PATH |
| Genewise, fastaseqfromGFF.pl i T-coffee | $ export PATH=/cursos/BI/bin:$PATH |
| Genewise | $ export WISECONFIGDIR=/cursos/BI/soft/wise-2.2.0/wisecfg |
3.1. Cerca de les proteïnes mitjançant BLAST:
En primer lloc, interessa trobar quines regions del genoma de Myotis davidii són susceptibles de contenir selenoproteïnes. Per saber-ho, es fa un blast de les selenoproteïnes obtingudes (veure obtenció de queries) contra el nostre genoma d'estudi.
BLAST (Basic Local Alignment Search Tool), és un programa informàtic que alinea localment una seqüència problema, o query, amb seqüències provinents d'una base de dades. Per tal de realitzar l'alineament, el programa segueix un algoritme heurístic que selecciona ràpidament les seqüències (hits) amb més homologia respecte la seqüència problema.
tBLASTn permet comparar una seqüència proteica (la nostra query) amb una base de dades de nucleòtids (genoma). tBLASTn tradueix totes les seqüències nucleotídiques en els 6 possibles ORFs (Open Reading Frames o marcs de lectura) per tal de comparar-les amb la seqüència problema i finalment mostrar les homologies trobades a partir de hits.
Per tal de realitzar-ho hem utilitzat la comanda següent:
| blastall -p tblastn -i query.fa -d /cursos/BI/genomes/project_2014/Myotis_davidii/genome.fa -o fitxerdesortida.fa |
| -p: tipus de blast; -i: ubicació de la query; -d: genoma del panda; -o: output |
L'output del tBLASTn és un llistat dels scaffolds trobats. Aquests indiquen les regions del genoma que tenen alta similitut amb la seqüència problema o query. La significança es pot valorar amb l'e-value present a cada scaffold. Aquest paràmetre descriu el nombre d'scaffolds que es poden esperar únicament per atzar quan realitzem l'anàlisi amb BLAST, i és significatiu quan és equivalent o inferior a 10-4.
3.2. Extracció de la regió genòmica d'interès:
A continuació, s'ha d'extreure la regió genòmica on l'alineament amb el tBLASTn indicava que es podria trobar la selenoproteïna. Abans d'això, s'ha d'indexar el genoma i a continuació extreure els scaffolds significatius. Finalment cal extreure la regió genòmica on es troba el nostre gen d'interès localitzat dins l'scaffold.
3.2.1. Realització de l'índex del nostre genoma:
Amb la finalitat d'indexar el genoma del nostre animal, hem utilitzat la comanda següent:
| $ fastaindex /cursos/BI/genomes/project_2014/Myotis_davidii/genome.fa sortida.index |
| cursos/BI/genomes/project_2014/Myotis_davidii/genome.fa: ubicació del genoma sortida.index : fitxer de sortida |
3.2.2. Extracció dels scaffolds estadísticament significatius:
Per extreure els scaffolds on es troba cadascun dels gens estadísticament significatius obtinguts amb el tBLASTn utilitzem el fastafetch mitjançant la següent comanda:
| $ fastafetch /cursos/BI/genomes/project_2014/Myotis_davidii/genome.fa sortida.index nomseq > nomseq.fa |
| cursos/BI/genomes/project_2014/Myotis_davidii/genome.fa: ubicació del genoma nomseq: nom de la regió que volem extreure (scaffold amb e-value per sota 10-4) nomseq.fa: arxiu que extraiem |
3.2.3. Extracció de la regió genòmica on es troba el gen a comparar d'interès:
Amb l'objectiu d'extreure el gen sencer localitzat en l'scaffold, seleccionem una regió que envolta el gen amb 50.000 nucleòtids per davant i 50.000 per darrere. D'aquesta manera ens assegurem d'incloure l'element SECIS de la proteïna. A fi d'aconseguir-ho hem utilitzat el programa fastasubseq mitjançant la comanda següent:
| $ fastasubseq nomseq.fa start length > genomic.fa |
| nomseq.fa: resultat del fastafech start: posició del primer nucleòtid del genoma del Myotis davidii que s'alinia amb la query en el tBLASTn – 50.000 nucleòtids length: 100.000 nucleòtids genomic.fa: arxiu de sortida |
4. Anotació genòmica:
4.1. Exonerate:
El següent a realitzar és assegurar-nos que el nostre hit es troba dins d'un exó, i que per tant, codifica per una proteïna. Per això, es realitzen les anotacions dels gens que estem buscant amb Exonerate, alineant el fragment de DNA que hem extret amb Fastasubseq amb la seqüència de DNA de la proteïna inicial. Fem servir la comanda:
| $ exonerate -m p2g --showtargetgff (--exhaustive) -q fitxerquery.fa -t fastasubseq.fa > exonerate.gff |
| -m p2g: model d’alineament (en aquest cas una proteïna com a query contra un genoma) showtargetgff: inclou el resultat en format GFF al fitxer de sortida --exhaustive: Fa una predicció exhaustiva, mostrant alineaments subòptims (comanda opcional) -q: ubicació de la query -t: indica la subseqüència obtinguda anteriorment sortida.gff: ubicació del fitxer de sortida en format gff on es representa l'alineament entre la query i la regió extreta amb el fastasubseq |
En alineaments mès pobres, usàvem la comanda "--exhaustive", que tot i fer anar el programa més lentament, pot trobar algún alineament que no s'ha trobat al fer l'exonerate normal
En aquest punt cal mirar si la X de la query està alineada amb un asterisc, cosa que indicaria un codó STOP o més probablement, en aquest cas, la presència d'una selenocisteïna.
4.1.1. Extracció dels exons:
Per tal d’extreure la seqüència exònica del fitxer anterior (exonerate.gff) utilitzem FastaseqfromGFF.pl amb la comanda següent, la qual ens indicarà les posicions dels exons:
| $ egrep -w exon sortida.gff > cDNA.gff |
| egrep: selecciona les línies on aparegui el patró definit -w exon: patró que volem buscar (exons) sortida.gff: fitxer obtingut del exonerate cDNA.gff: fitxer de sortida que indica les posicions dels exons. |
Una vegada creat el fitxer cDNA.gff ja podem fer servir el programa fastaseqfromGFF.pl, el qual aplicarà la informació a la seqüència del fastasubseq per tal d'obtenir el DNA dels exons:
| $ fastaseqfromGFF.pl genomic.fa cDNA.gff > cDNA.fa |
| genomic.fa: arxiu extret mitjançant fastasubseq cDNA.gff: arxiu que conté els exons del hit extret cDNA.fa: és el fitxer que contindrà en format fasta la seqüència de DNA del cDNA.gff |
4.1.2. Traducció dels exons de DNA a proteïna:
Un cop tenim els exons, ens interessa buscar la proteïna per la qual codifiquen, és a dir, volem traduir la seqüència exònica. Per realitzar-ho hem fet servir el programa fastatranslate, el qual proporciona un fitxer amb la traducció en sis marcs de lectura possibles (ORFs). La comanda emprada és la següent:
| $ fastatranslate cDNA.fa > aa_gen.mfa |
| cDNA.fa: ubicació del cDNA aa_gen.mfa: fitxer de sortida en format multifasta que conté els 6 ORFs |
El següent pas és fer el T-coffee (explicat en el següent apartat), però per dur-lo a terme hem de seleccionar prèviament la millor traducció dels 6 ORFs. Manualment, hem buscat quina seqüència s'assemblava més a la nostra query i hem introduït la següent comanda:
| fastatranslate -F X cDNA.fa > aa_gen_1.mfa |
| X: indica ORFs seleccionats que pot ser en reverse (-3, -2, -1) o en forward (1, 2, 3) cDNA.fa: ubicació del cDNa aa_gen_1.mfa: fitxer de sortida en format multifasta que conté només 1 ORF |
4.2. T-COFFEE
El T-COFFEE (Tree-based Consistency Objective Function for alignment Evaluation) és un programa que permet fer l'alineament global de la proteïna resultant en el fastatranslate amb la proteïna problema extreta de la base de dades (query). El resultat indicarà presència o absència d'homologia entre les dues seqüències. La comanda utilitzada és:
| $ t_coffee query.fa gen_aa_1.mfa > t_coffee.fa |
| query.fa: proteïna extreta de la base de dades gen_aa_1.mfa: proteïna obtinguda del fastatranslate (exonerate) t_coffee.fa: fitxer de sortida |
4.3. Genewise
El programa Genewise genera una nova anotació del gen, utilitzant però, un algorisme diferent a l'Exonerate. Per aquest motiu, el Genewise ens ajuda a contrastar la informació que hem obtingut amb l'Exonerate. La comanda és la següent:
| $ genewise -pep -pretty -cdna -gff -both query.fa genomic.fa > sortida.gff |
| -pep: mostra al fitxer de sortida la seqüència peptídica predita -pretty: mostra l’alineament -cdna: mostra la seqüència genòmica alineada -gff: resultat en format gff query.fa: ubicació de la nostra query
genomic.fa: ubicació del resultat del fastasubseq sortida.gff: fitxer de sortida |
5. Automatització
Per automatitzar el procés d'anotació, hem creat un programa en bash per fer els exports i un programa en perl per fer la resta de passos descrits anteriorment.
5.1. Definitiu.sh
Per a poder fer els exports necessaris per a que el programa funcioni, vam tenir que crear un petit programa en bash (donat que les ordres són ordres d'export, que s'executen al shell) que fa els exports i crida al programa definitiu.pl. Per al bon funcionament del programa, ha d'estar dins la carpeta "luke" juntament amb el programa definitiu.pl i la query. El programa definitiu.sh podeu trobar-lo aquí.
5.2. Definitiu.pl
El programa definitiu.pl fa l'anotació d'una proteïna al genoma de Myotis davidii, per exonerate i genewise, a part de fer el tcoffee. El programa s'executa tot sol en executar el programa en bash, ja que necessita els exports. El programa analitzarà, donada una query, si aquesta es troba en el genoma de Myotis davidii. Per al bon funcionament del programa, ha d'estar en la carpeta "luke", juntament amb el programa definitiu.sh i la query. El programa definitiu.pl podeu trobar-lo aquí. Dins del programa, podeu trobar algunes anotacions sobre el funcionament de cada pas.
6. Cerca d'elements SECIS:
Per predir els elements SECIS hem utilitzat el programa online Seblastian, el qual es pot utilitzar tant per identificar selenoproteïnes ja conegudes com per predir noves selenoproteïnes.
Seblastian utilitza SECISearc3 (versió més nova que SECISearch) per detectar elements SECIS, i posteriorment prediu la seqüència de la selenoproteïna codificada upstream dels elements SECIS.
Per gaudir del seu servei, hem pujat el document de sortida del fastasubseq (genomic.fa) i n'hem extret l'output.