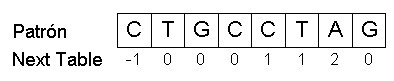| Introducción | Objetivos | Metodología | Programas | Resultados | Conclusiones | Referencias | Autoras | Agradecimientos |
METODOLOGÍA
Para llevar a cabo el proyecto y abordar el problema de la correspondencia exacta entre símbolos, hemos tenido que implementar dos algoritmos: el algoritmo simple de correspondencia exacta y el algoritmo de Knuth-Morris-Pratt. |
|||||||||||||||||
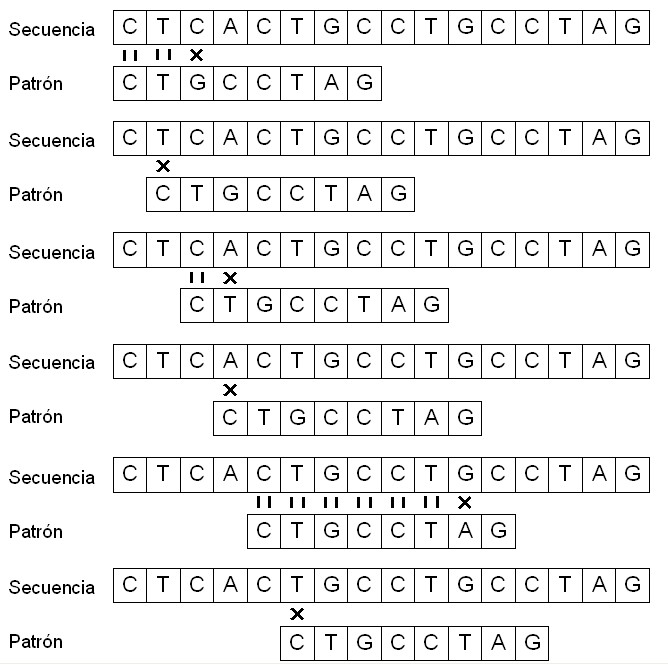
Este algoritmo se basa en buscar patrones exactos dentro de una secuencia de DNA, por medio del algoritmo "tonto" de recorrer toda la secuencia nucleótido por nucleótido, haciendo la comparación oportuna con el patrón.El principal factor a tener en cuenta es el problema de encontrar todos los posibles desplazamientos a lo largo de la secuencia.
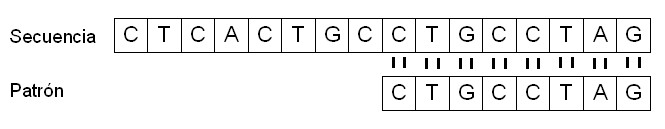
Por ejemplo, si consideramos una secuencia t = {CTCACTGCCTGCCTAG} y un patrón p = {CTGCCTAG}, el patrón p se encuentra dentro de t a partir del noveno símbolo, y podemos decir que el patrón p aparece en t con un desplazamiento s, en este caso, s = 8.
Esto hace que los símbolos:
|
|||||||||||||||||
|
t[s+1]t[s+2]t[s+3]...t[s+m] |
|||||||||||||||||
sean exactamente los de p, es decir: |
|||||||||||||||||
t[s+1] == p[0] t[s+2] == p[1] t[s+3] == p[2] ... t[s+m] == p[m-1] |
|||||||||||||||||
Así, podremos llegar a encontrar un máximo de n - m + 1 desplazamientos s.
Este algoritmo, también conocido como de "fuerza bruta", funciona de una manera simple y obvia: después de un mismatch entre patrón y secuencia, el proceso de comparación se detiene y se incia otra vez desplazando el patrón una posición sobre la secuencia. En el ejemplo, la comparación entre patrón y secuencia comienza en la primera posición de ambos. Como los símbolos son iguales, la comparación continúa con los dos nucleótidos siguientes, en los que también hay coincidencia. En este caso, la comparación también continúa con los dos nucleótidos siguientes pero no se produce coincidencia. Así pues, la comparación entre patrón y secuencia empieza otra vez, pero ahora se compara la posición 0 del vector con la posición 1 del vector secuencia (segundo nucleótido), i así se hace sucesivamente, desplazando el inicio de la comparación del patrón sobre la secuencia únicamente un nucleótido.
|
|||||||||||||||||
|
|||||||||||||||||
Como resultado, encontramos que la secuencia t contiene el patrón p con un desplazamiento s igual a 8. |
|||||||||||||||||
|
|||||||||||||||||
|
|
|||||||||||||||||
 ALGORITMO DE KNUTH-MORRIS-PRATT ALGORITMO DE KNUTH-MORRIS-PRATT
El algoritmo de Knuth-Morris-Pratt es un algoritmo lineal para la búsqueda exacta de patrones. Es parecido al algoritmo simple de correspondencia exacta pero la principal diferencia es que el algoritmo de Knuth-Morris-Pratt se basa en el hecho que, después de encontrar el primer símbolo que no coincide entre el patrón y la secuencia, nosotros ya conocemos los símbolos del patrón que hemos comparado hasta ese punto. De esta manera, se consigue que después de un emparejamiento incorrecto el proceso no continúe (desde el inicio del patrón) con el siguiente carácter de la secuencia, y así no tener que comparar posiciones que ya se sabe previamente que no darán lugar a una ocurrencia exacta del patrón.
El KMP se beneficia de la información que el propio patrón contiene ya que se desconoce la secuencia donde se realiza la búsqueda. Para aprovechar este hecho, hay que construir una tabla de desplazamientos coincidentes dentro del patrón antes de empezar la búsqueda. Esta tabla, llamada next table, tiene una posición para cada posición del patrón, y en cada posición n se introducirá el número máximo de símbolos coincidentes con el patrón antes de esa posición (sin coincidir el prefijo entero de n símbolos). Es decir, en cada posición n se comprueban las coincidencias existentes entre los diferentes prefijos de n y el patrón que precede a n.
Continuando con el ejemplo anterior en que teníamos una secuencia t ={CTCACTGCCTGCCTAG} y un patrón p = {CTGCCTAG}, y el patrón p se encuentra dentro de t a partir de noveno símbolo. Entonces, como antes, podemos decir que el patrón se encuentra en t con un desplazamiento s = 8.
En el KMP, primero de todo se calcula la next table para el patrón. Para el patrón p = {CTGCCTAG} sería la siguiente:
|
|||||||||||||||||
|
|||||||||||||||||
A la hora de construir la next table, se asigna un valor de -1 a la posición 0 del vector de la next table y un 0 a la posición 1. El valor -1 en la posición 0 es una señal cualquiera que nos sirve para comenzar la comparación con el siguiente nucleótido de la secuencia en caso que se produzca un mismatch entre la posición 0 del vector patrón y un símbolo de la secuencia. En la posición 1 del vector de la next table se tendría que comparar el prefijo de la posición 1, formado exclusivamente por la posición 0, con el patrón antes de la posición 1. Como el prefijo, aunque es coincidente, siempre será máximo, asignaremos directamente el valor 0 a la posición 1. Continuamos con la posición 2 del vector patrón, donde se mira el número máximo de nucleótidos coincidentes con el patrón antes de esa posición. En este caso es sencillo, ya que sólo hemos de mirar si la posición 1 (T) es igual que la posición 0 (C) del vector patró. Como no son iguales, el valor del vector de la next table para la posición 2 será 0. Si pasamos a la posición 3, hemos de comparar si el prefijo formado por las posiciones 1 i 2 o el formado únicamente por la 2 coinciden con el patrón des de su inicio, es decir, si el prefijo constituído por las posiciones 1 y 2 es igual a las posiciones 0 y 1 o si la posición 2 es igual que la posición 0 del patrón. En este caso, no se produce ninguna coincidencia, de manera que el valor de la next table para la posición 3 es 0. Si miramos la posición 4, tendremos que comparar los prefijos formados por las posiciones 1, 2 y 3, por las posiciones 2 i 3 o por la posición 3 con el patrón que les precede. Aquí vemos que la posición 3 es igual a la posición 0 del vector patrón, así que el valor del vector de la next table para la posición 4 es igual a 1 (el prefijo mayor en la posición 4 que coincide con el patrón está formado por un nucleótido). Si ahora nos centrado en la posición 5, ocurre lo mismo que con la posición 4, y el prefijo mayor que coincide con el patrón es de un nucleótido, de manera que el valor de la next table para la posición 5 es 1. Cuando miramos la posición 6, tenemos diferentes prefijos: los formados por las posiciones 1, 2, 3, 4 y 5, por 2, 3, 4 y 5, por 3, 4 y 5, por 4 y 5 y por 5. En este caso, el prefijo formado por las posiciones 4 y 5 coinciden con las posiciones 0 y 1 del vector patrón, y pondremos un 2 (2 nucleótidos coincidentes) en la posición 6 del vector de la next table. Para finalizar con la next table de este patrón, miramos la última posición, la 7. Ninguno de los prefijos para esta posición coincide con el patrón. Así pues, el vector de la next table tendrá un valor de 0 en la posición 7.
|
|||||||||||||||||
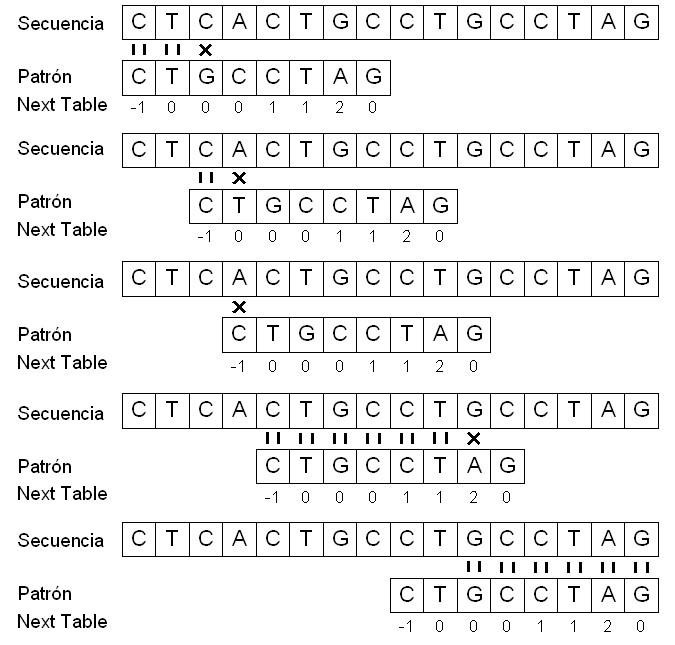
Una vez construída la next table, se procede a implementar el KMP. El funcionamiento de este seria el siguiente: siempre que hay una correspondencia entre dos símbolos (uno de la secuencia y el otro del patrón) se pasa a la siguiente posición, tanto de la secuencia como del patrón. En cambio, cuando tenemos dos símbolos diferentes, se empieza a buscar el patrón en la secuencia a partir de la posición del patrón que nos indica la next table.
Por ejemplo:
En la penúltima comparación del patrón con la secuencia, en que el patrón empieza a comparar con el nucleótido 5 de la secuencia, se produce una correspondencia entre los símbolos hasta llegar al nucleótido número 11 de la secuencia, donde hay una G, que se compara con la A del patrón. Como se produce este mismatch, miramos el valor de esta A en la next table y observamos un 2. Entonces, se empieza a comparar el símbolo el símbolo G de la secuencia con el de la posición 2 del vector patrón. Finalmente, encontramos el patrón p dentro de la secuencia t con un desplazamiento s igual a 8.
|
|||||||||||||||||
|
|||||||||||||||||
|
|
|||||||||||||||||
Una vez implementado el algoritmo de Knuth-Morris-Pratt en lenguaje de programación Perl para la búsqueda de subsecuencias únicas en el genoma humano de los genes GH, IGF-1 y EPO, se determinó que el tiempo de ejecución del programa sería de unos 40* días aproximadamente para cada gen, en el mejor de los casos. Este tiempo es muy superior a los antecendentes de que disponíamos por parte del supervisor del trabajo, el Dr. Robert Castelo, que ya había implementado el algoritmo simple de correspondencia exacta en lenguaje C con la finalidad de encontrar patrones únicos del gen de la EPO de ratón en el genoma de este organismo. En un principio, se había pensado que implementando el algoritmo de Knuth-Morris-Pratt, que es más eficiente que el algoritmo simple de correspondencia exacta, habría una ganancia computacional, aunque el lenguaje Perl es más lento que el lenguaje C. Pero la gran diferencia en términos de tiempo entre estos dos lenguajes de programación ha hecho imposible obtener las subsecuencias únicas de los genes de interés en el periodo establecido. No obstante, hemos creado un programa basado en la implementación del algoritmo de Knuth-Morris-Pratt que es capaz de realizar esta búsqueda de patrones únicos en todo el genoma dado un mRNA. Para la comprobación del correcto funcionamiento del programa hemos corroborado los resultados obtenidos por el Dr. Robert Castelo, es decir, comprobar que los tres patrones únicos encontrados en el gen de la EPO de ratón son subsecuencias únicas del cromosoma 5 del genoma de ratón, donde se encuentra el locus de este gen.
* El cálculo de estos 40 días aproximados se llevó a cabo de la siguiente forma:
|
|||||||||||||||||
|
|
|||||||||||||||||
Tal como hemos comentado en el apartado de objetivos, para poder monitorizar la expresión génica del mRNA en el caso de un posible dopaje genético, hace falta disponer de secuencias específicamente marcadas y que sean capaces de hibridar selectivamente con el mRNA diana. Por este motivo, interesa obtener unos patrones únicos de los genes de interés (GH, IGF-1 y EPO) que sean accesibles a los PNAs marcados y no estén escondidos en la estructura secundaria del mRNA. Así pues, una vez obtenidos los patrones únicos en el genoma (concretamente en el cromosoma 5 del genoma de ratón) a través de la implementación del algoritmo de Knuth-Morris-Pratt, se predice la estructura secundaria del mRNA y la localización de las las subsecuencias encontradas del gen de la EPO con el programa mFOLD. Este programa, creado por el Dr. Michael Zuker del Rensselaer Polytechnic Institute's School of Science y disponible como servicio web, es un software que predice el plegamiento y la estructura secundaria tanto de RNA como de DNA. |
|||||||||||||||||
|
|
|||||||||||||||||
La metodología seguida en este proyecto es la siguiente:
Las secuencias utilizadas para la elaboración de este proyecto, obtenidas en la base de datos de NCBI, son las siguientes:
|