EXPLICACIÓ DETALLADA DEL SCRIPT
 | EXPLICACIÓ DETALLADA DEL SCRIPT |
Per a començar, vam crear un petit programa en perl que llegia les coordenades de les regions ENCODE i treia per pantalla aquelles anotacions que se solapaven amb uns fitxers de gens en format goldenPath ( link a mètodes ) que contenien les anotacions de gens de RefSeq, Ensembl, genscan i geneid. Com el programa consumia molta memòria, vam considerar que el primer que calia fer era dividir els fitxers de dades originals en els diferents cromosomes que contenien. Per a realitzar aquesta tasca les comandes que vam utilitzar van ser les següents:
bash$ egrep "chr19" /disc8/genomes/H.sapiens/golden_path_200307/refGene.txt > RefSeq19.txt
bash$ egrep "chr19" /disc8/genomes/H.sapiens/golden_path_200307/ensGene.txt > Ensembl19.txt
bash$ egrep "chr19" /disc8/genomes/H.sapiens/golden_path_200307/genscan.txt > genscan19.txt
bash$ egrep "chr19" /disc8/genomes/H.sapiens/golden_path_200307/geneid.txt > geneid19.txt
En el cas del cromosoma 1 i 2 l'ordre era la següent: bash$ egrep "chr1[^0-9]" /disc8/genomes/H.sapiens/golden_path_200307/refGene.txt > RefSeq1.txt
I aquestes mateixes ordres canviant cada vegada el cromosoma fins arribar fins l'X.
Ara ja si que tenim els fitxers llestos per a aplicar el programa. A continuació passarem a descriure breument el cos del programa. Si voleu veure la completa versió d'aquest feu un click.

1. Explicació del programa
En primer lloc declarem les variables:
my @cromosoma; my @inici; my @final; my $i = 0; my $notrobat = 0; i apliquem la composició iterativa While mentre hi hagi línies que encaixin amb l'expressió regular:
$_ =~m/\A(\w+)\s+(\d+)\s+(\d+)/; dels gens que estàn anotats amb el format ENCODE.txt
chr1 148374642 128874642 ENr231
Aleshores capturem les variables:
$cromosoma[$i] = $1; on capturem el nom del cromosoma de ENCODE.txt $inici[$i] = $2; on capturem l'inici de transcripció d'ENCODE.txt $final[$i] = $3; on capturem el final de transcripció d'ENCODE.txt i anem sumant cada posició $i = $i +1; i un cop ha acabat tanquem el fitxer ENCODE.
Declarem unes noves variables:
my @id; my @chr; my @stra; my @itrans; my @ftrans; my @itrad; my @ftrad; my @numex; my @exon; my $posicio = 0;
i així anem obrint els diferents fitxers de cromosomes per les 4 diferents bases de dades: Ensembl, RefSeq, Geneid i Genscan amb una composició iterativa While mentre hi hagi línies que encaixin amb l'expressió regular
$_= ~m/(\w+)\s+(\w+)\s+([\+\-])\s+(\d+)\s+(\d+)\s+(\d+)\s+(\d+)\s+(\d+)\s+([\d+\,\s]+)/; que estan en format golden_path:
- RefSeq: NM_014188 chr1 - 1428455 1461406 1428847 1461339 5
1428455,1430650,1431644,1451554,1461259, 1428949,1430769,1431784,1451698,1461406,
- Ensembl: ENST00000326347 chr1 - 25000 25357 25000 25357 2
25000,25139, 25037,25357,
- Genscan: NT_011387.6 chr20 + 298788 329279 298788 329279 6
298788,299911,301670,311652,325281,328853, 298855,300084,301891,311722,325476,329279,
- Geneid: chr1_18.1 chr1 - 1052992 1055565 1052992 1055565 6
1052995,1053141,1053379,1054075,1055100,1055420, 1053063,1053270,1053576,1054142,1055223,1055565,
és a dir, If (!open(CROMOSOMES," RefSeqX.txt")) és aquí on anem canviat el nom del fitxer i el nom del cromosoma.
Aleshores capturem aquestes variables:
$id[$posicio]= $1; on capturem l'identificador del gen $chr[$posicio] = $2; on capturem cada cromosoma $stra[$posicio] = $3; on capturem el strand (+/-) $itrans[$posicio] = $4; on capturem inici transcripció $ftrans[$posicio] = $5; on capturem final transcripció $itrad[$posicio] = $6; on capturem inici traducció $ftrad[$posicio] = $7; on capturem final traducció: $numex[$posicio] = $8; on capturem en nombre d'exons @exon = $9=~m/(\d+)/g; on capturem en un vector els diferents valors dels exons
i finament, es fa la comparació per cada cromosoma diferent del fitxer d'ENCODE:
if($cromosoma[$i] eq 'chrX '){ és aquí on canviem el nom de cada cromosoma
if (($itrans[$posicio] >= $inici[$i]) && ($ftrans[$posicio] <= $final[$i])){ i printem la solapació dels diferents fitxers de cromosomes per a les 4 bases de dades en format Golden_Path:
print "$id[$posicio] $chr[$posicio] $stra[$posicio] $itrans[$posicio] $ftrans[$posicio] $itrad[$posicio] $ftrad[$posicio] $numex[$posicio]"; Si no hi ha cap solapació entre ENCODE.txt i els diferents fitxers de cromosomes pes les 4 bases de dades en format Golden_Path, printem:
print "No s'ha trobat valors dins de ENCODE dins el RefSeq del chrX\n"; és aquí on canviem el nom de cada cromosoma no solapat.
2. Desenvolupament del treball
Els resultats d'aplicar el progama en el diferents fitxers golden_path de cada base de dades (RefSeq, Ensembl, geneid i genscan), els vam redireccionar a quatre fitxers de text on es trobaven les notacions dels cromosomes, les coordenades d'inici i final de trancripció dels quals, se solapaven amb les de ENCODE.txt.
Per a fer aquesta tasca vam utilitzar les següents comandes:
bash$ ./RefSeq.pl > RefSeqEN.txt per a redireccionar el resultat del primer cromosoma
bash$ ./RefSeq.pl >> RefSeqEN.txt per redireccionar la resta de cromosomes.
Aquesta operació la vam utilitzar per a les quatre bases de dades obtenint els següents fitxers de text:
RefSeqEN.txt
Un cop fet això, vam contar quantes anotacions respecte els fitxers de dades originals havíem obtingut:
bash$ egrep "NM" /disc8/.../refGene.txt | wc
bash$ egrep "ENS" /disc8/.../ensGene.txt | wc
bash$ egrep "NT" /disc8/.../genscan.txt | wc
bash$ egrep "chr[0-9]_" /disc8/.../geneid.txt | wc
i també vam obtenir el número d'anotacions dels fitxers de dades originals de RefSeq, Ensembl, genscan i geneid, respectivament
bash$ egrep "NM" RefSeqEN.txt | wc
bash$ egrep "ENS" EnsemblEN.txt | wc
bash$ egrep "NT" genscanEN.txt | wc
bash$ egrep "chr[0-9]_" geneidEN.txt | wc
Els resultats d'aquestes comandes es mostren a la següent taula:
| NÚMERO D'ANOTACIONS | |||
| disc8/.../refGene.txt | 21.094 | RefSeqEN.txt | 442 |
|---|---|---|---|
| disc8/.../ensGene.txt | 27.937 | EnsemblEN.txt | 551 |
| disc8/.../genscan.txt | 42.972 | genscanEN.txt | 585 |
| disc8/.../geneid.txt | 16.128 | geneidEN.txt | 161 | TOTAL | 108.131 | 1.739 |
El següent pas, és ajuntar els fitxers RefSEqEN.txt amb EnsemblEn.txt i geneidEn.txt amb genscanEN.txt; per a fer-ho simplement fem el redireccionament que es mostra a continuació:
bash$ EnsemblEN.txt >> RefSeqEN.txt
bash$ geneidEN.txt >> genscanEN.txt
Per tant, ara en el fitxer RefSeqENj.txt tenim la informació conjunta de RefSeqEN.txt i EnsemblEN.txt i a genscanENj.txt tenim la informació conjunta de geneidEN.txt i genscanEN.txt. Ara el número d'anotacions d'aquest dos fitxers és el següent:
| NÚMERO D'ANOTACIONS | |||||
| disc8/.../refGene.txt | 21.094 | RefSeqEN.txt | 442 | RefSeqENj.txt | 993 |
|---|---|---|---|---|---|
| disc8/.../ensGene.txt | 27.937 | EnsemblEN.txt | 551 | ||
| disc8/.../genscan.txt | 42.972 | genscanEN.txt | 585 | genscanENj.txt | 746 |
| disc8/.../geneid.txt | 16.128 | geneidEN.txt | 161 | ||
| TOTAL | 108.131 | 1.739 | 1.739 | ||
On 993 no és res més que la suma dels 442 + 551 de ReSeqEN.txt i EnsemblEN.txt i 746 la suma de 586 + 161 de genscanEN.txt i geneidEN.txt
Ara amb aquests diferents passos ja hem completat el pas 1 i 2 de mètodes
Ara passem al tercer punt del treball, on dividirem el fitxer RefSeqENj.txt en diferents fitxers on cada un tingui les anotacions de RefSeq i Ensembl per a cada cromosoma. Farem el mateix per a les anotacions de genscanENj.txt.
Les comandes utilitzades són les que es mostren:
bash$ egrep "chr19" RefSeqENj.txt > RS19.txt
bash$ egrep "chr19" genscanENj.txt > gege19.txt
En el cas del cromosoma 1 i 2 l'ordre era la següent: bash$ egrep "chr1[^0-9]" RefSeqENj.txt > RS1.txt
D'aquesta manera obtenim un conjunt de fitxers RS.txt i gege.txt per a cada cromosoma diferent. Sobre tots aquests fitxers apliquem el següent conjunt d'ordres:
bash$ gawk '{printf "%s\t%s\t%s\t%s\t%s\t%s\t%s\t%s\t",$1,$2,$3,$4,$5,$6,$7,$8;for(i=9;i<9+$8;i++){printf "%s",$i}printf "\t";for(i=9+$8;i<=9+$8*2;i++){printf "%s",$i}printf "\n"}' RS1.txt | sort -k9 -k10 | uniq -f 8 > RSjuntfi.txt
bash$ gawk '{printf "%s\t%s\t%s\t%s\t%s\t%s\t%s\t%s\t",$1,$2,$3,$4,$5,$6,$7,$8;for(i=9;i<9+$8;i++){printf "%s",$i}printf "\t";for(i=9+$8;i<=9+$8*2;i++){printf "%s",$i}printf "\n"}' gege1.txt | sort -k9 -k10 | uniq -f 8 > gegejuntfi.txt
Fins a la comanda sort el que es fa és donar un determinat format als diferens fitxers: a la sortida del programa els inicis i finals d'exons estaven tots separats per espais, per tant, cada inici i cada final era un field diferent. Això complica les coses alhora de treballar amb les comandes del shell (sort, uniq...) per tant, el que fem és ajuntar tots els inicis d'exons en un únic camp($9) i els finals d'exons en un altre camp ($10):
Abans de la comanda: NT_029419.30 chr12 - 38772889 38811110 38772889 38811110 3
38772889, 38785321, 38810987, 38773002, 38785658, 38811110Després de la comanda: NT_029419.30 chr12 - 38772889 38811110 38772889 38811110 3
38772889,38785321,38810987, 38773002,38785658,38811110
Amb el sort -k el que aconseguim és que s'ordenin només els camps $9 i $10, que corresponen als inicis i finals d'exons. Amb l'uniq -f el que aconseguim és eliminar les repeticions només als camps $9 i $10, perquè els altres camps s'ignoren. No cal dir, que aquests passos els fem tant pels fitxres RS.txt i gege.txt
Com a resultats obtenim els fitxers RSjuntfi.txt i gegejuntfi.txt amb les anotacions dels gens que s'han solapat entre RefSeq i Ensembl i geneid i genscan.
| NÚMERO D'ANOTACIONS | |||||||
| disc8/.../refGene.txt | 21.094 | RefSeqEN.txt | 442 | RefSeqENj.txt | 993 | RSjuntfi.txt | 924 |
|---|---|---|---|---|---|---|---|
| disc8/.../ensGene.txt | 27.937 | EnsemblEN.txt | 551 | ||||
| disc8/.../genscan.txt | 42.972 | genscanEN.txt | 585 | genscanENj.txt | 746 | gegejuntfi.txt | 746 |
| disc8/.../geneid.txt | 16.128 | geneidEN.txt | 161 | ||||
| TOTAL | 108.131 | 1.739 | 1.739 | 1670 | |||
Ara que ja tenim aquests fitxers el redireccionem tots dos a un que tindrà els quatre tipus d'anotacions de les diferents bases de dades: juntfi.txt.Per a fer-ho hem fet el següent:
bash$ RSjuntfi.txt > juntfi.txt
bash$ gegejuntfi.txt >> juntfi.txt
Un cop fet això tornem a dividir aquest fitxer en els diferents cromosomes, per variar:
bash$ egrep "chr19" juntfi.txt > juntfi19.txt i així per a tots els cromosomes
El següent pas és fer el mateix que en el pas 1 i 2: utilitzant les comandes sort, uniq...mirar aquelles anotacions que se solapen
( annex1 ) A més a més, per a restringir més aquest solapament, restarem les posicions inici de transcripció de gen ièssim - fi de transcripció de gen ièssim de gens consecutius. A continuació es mostra un esquema per a poder entendre millor aquest plantejament:
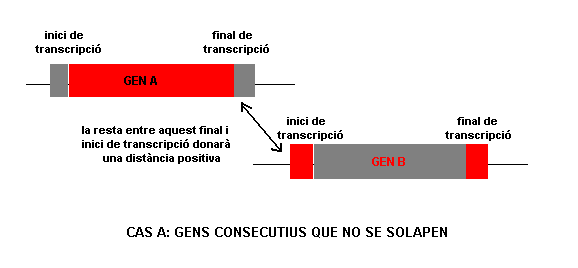
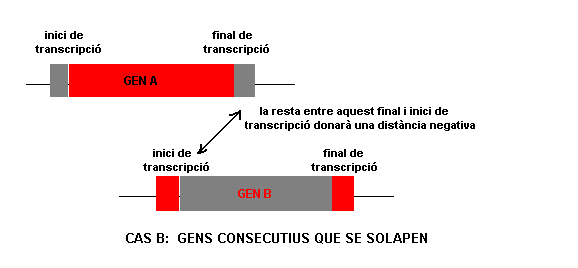
La comanda utilitzada es mostra a continuació:
bash$ gawk '{print $1,$2,$3,$4,$5,$6,$7,$8,$9,$10,$4-b;b=$5}'juntfi.txt | sort -k9 -k10 | uniq -f 8 > juntfisuma.txt
Ara hem aconseguit tenir el fitxer juntfisuma.txt, on en el field $11 tenim el resultat de la resta anterior. Aquest valor de $11, pot ser tant positiu com negatiu; els valors positius indiquen que les anotacions no se solapen i el negatius que sí que se solapen. A nosaltres ens interessen els valors que no se solapen (per tant, els positius) per a poder passar al punt 4 de mètodes .
Les anotacions que donen a la resta un resultat negatiu les separem i les enviem a un fitxer de text de la seguent manera:
bash$ egrep "\-[0-9]"juntfisuma > solapamentfi.txt
Així a solapamentfi.txt tindrem les anotacions dels gens que se solapen totalment entre ells, o bé els que alguna part d'ells queda compresa entre els inicis i finals de transcripció d'un altre gen. Si contem quantes anotaicons ens queden amb la següent comanda obtenim la taula 4:
bash$ egrep "NM" solapamentfi.txt | wc
bash$ egrep "ENS" solapamentfi.txt | wc
bash$ egrep "NT" solapamentfi.txt | wc
bash$ egrep "chr[0-9]_" solapamentfi.txt | wc
| NÚMERO D'ANOTACIONS | |||||||||
| disc8/.../refGene.txt | 21.094 | RefSeqEN.txt | 442 | RefSeqENj.txt | 993 | RSjuntfi.txt | 924 | solapamentfi.txt | 259 |
|---|---|---|---|---|---|---|---|---|---|
| disc8/.../ensGene.txt | 27.937 | EnsemblEN.txt | 551 | solapamentfi.txt | 408 | ||||
| disc8/.../genscan.txt | 42.972 | genscanEN.txt | 585 | genscanENj.txt | 746 | gegejuntfi.txt | 746 | solapamentfi.txt | 314 |
| disc8/.../geneid.txt | 16.128 | geneidEN.txt | 161 | solapamentfi.txt | 129 | ||||
| TOTAL | 108.131 | 1.739 | 1.739 | 1670 | 1.110 | ||||
Fem un procediment semblant per tal de seleccionar les que no se solapen, és a dir, les que la suma dóna un número negatiu com a resultat:
Si contem quantes anotacions ens queden (a nosolapamentfi.txt) amb la següent comanda obtenim la taula 5:bash$ gawk '{if ($11>'0'){print $0}}'juntfisuma > nosolapamentfi.txt
bash$ egrep "NM" nosolapamentfi.txt | wc
bash$ egrep "ENS" nosolapamentfi.txt | wc
bash$ egrep "NT" nosolapamentfi.txt | wc
bash$ egrep "chr[0-9]_" nosolapamentfi.txt | wc
| NÚMERO D'ANOTACIONS | |||||||||||
| disc8/.../refGene.txt | 21.094 | RefSeqEN.txt | 442 | RefSeqENj.txt | 993 | RSjuntfi.txt | 924 | solapamentfi.txt | 259 | nosolapamentfi.txt | 114 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| disc8/.../ensGene.txt | 27.937 | EnsemblEN.txt | 551 | solapamentfi.txt | 408 | nosolapamentfi.txt | 143 | ||||
| disc8/.../genscan.txt | 42.972 | genscanEN.txt | 585 | genscanENj.txt | 746 | gegejuntfi.txt | 746 | solapamentfi.txt | 314 | nosolapamentfi.txt | 247 |
| disc8/.../geneid.txt | 16.128 | geneidEN.txt | 161 | solapamentfi.txt | 129 | nosolapamentfi.txt | 32 | ||||
| TOTAL | 108.131 | 1.739 | 1.739 | 1670 | 1.110 | 536 | |||||
Tal i com ens demana el punt 4 de mètodes per les anotacions de geneid i genscan que no se solapen hem de buscar amb ESTs evidència experimental de que aquests gens poden ser reals. Com d'anotacions de genscan en tenim 247 i de geneid en tenim 32, per manca de temps i per problemes que ja s'han comentat a ( l'annex1 ) només realitzarem aquest pas per a les seqüències de geneid; per tant, amb la comanda:
bash$ egrep "NT" nosolapamentfi.txt > final.txt
bash$ egrep "chr[0-9]_" nosolapamentfi.txt >> final.txt creem el fitxer final.txt on només hi ha les anotacions de geneid i genscan.
bash$ egrep "chr[0-9]_" final.txt > geneidfinal.txt D'aquesta manera separem les 32 anotacions de geneid (a geneidfinal.txt)que seran amb les que treballarem a partir l'ara.
Com la comprovació experimental de que aquestes anotacions són gens reals la fem a partir dels ESTs ( resultats ), hem de tenir en compte que només treballarem amb aquelles anotacions en les quals els ESTs s'aliniïn amb més d'un exó, ja que això reforçarà la hipòtesi que aquests gens predits siguin gens reals. Així doncs:
bash$ gawk '{if ($8 > 1){print ($5 - $4)/1000,$0}}'geneidfinal.txt > kbgeneidfinal.txt Amb el gawk reduïm les anotacions que tenen més d'un exó, de tal manera que el nombre final d'anotacions és 25. A més, fem que al fitxer kbgeneidfinal.txt apareguin el nombre de kilobases al field $1 de cada gen per tal de seguir amb el treball.
Observant el nombre de kilobases de kbgeneidfinal.txt veiem que hi ha 2 fitxers massa grans: un de 427,471 kb i 195,593 kb; aquests dos fitxers els eliminarem ja que no ens en fiem que puguin ser gens pel gran tamany. A més, sent tan grans també aumentarà la possibilitat de que s'aparellin més ESTs donant falsos positius. Per eliminar aquestes seqüències i quedar-nos amb 23, fem el següent:
bash$ sort -k 1 -n kbgeneidfinal.txt | gawk '{if ($1<'195.593'){print $0}}' > anotacions.txt De tal manera que primer ordenem el fitxer pel nombre de kilobases i després seleccionem aquelles més grans de 195,593 kb en el fitxer anotacions.txt.
Un cop fet això, ara el que calia era recuperar les sequencies genòmiques d'aquestes 23 anotacions; per a fer-ho tot amb un únic pas, vam utilitzar la següent comanda:
bash$ cat anotacions.txt | gawk '{printf"fastachunk/disc8/genomes/H.sapiens/golden_path_200307/chromFa_msk/%s.fa.masked %d %d |fold -60 > %s.fa\n", $2, $4-1000, $5-$4+2000,$1}'> agafarseqs.sh De tal manera, que primer donem un determinat format al fitxer (per exemple, amb el fold aconseguim que cada línia de seqüència ens quedi amb 60 unitas).
Per treballar amb aquestes seqüències necessitem emmascarar els elements repetitius per tal que no ens interfereixin amb les nostres predicions de gens ja que són regions no codificants; això ho fem amb la part de la comanda "chromFa_msk/%s.fa.masked". El fitxer resultant es veuria d'aquesta manera si fos en extensió .txt (anotacionsprograma.txt).
Ara executarem el programa fastachunk:
bash$ source anotacions.sh per obtenir les diferens seqüències genòmiques de cada cromosoma. Les seqüències en format fasta són:
chr1_1626.1.fa
chr1_1628.1.fa
chr2_1226.1.fa
chr2_2065.1.fa
chr2_2056.1.fa
chr2_2205.1.fa
chr5_1143.1.fa
chr5_977.1.fa
chr5_979.1.fa
chr6_928.1.fa
chr6_1127.1.fa
chr6_680.1.fa
chr6_923.1.fa
chr6_1129.1.fa
chr6_921.1.fa
chr7_1259.1.fa
chr7_1204.1.fa
chr7_292.1.fa
chr7_1261.1.fa
chr7_911.1.fa
chr7_1258.1.fa
chr7_1256.1.fa
chr9_1068.1.fa
Ara tenim 23 fitxers on a cadascun hi ha la seqüència del cromosoma en format FASTA.
A partir d'aquí treballarem amb diferents pàgines d'internet per tal de fer la predicció de futurs gens. Per una sèrie de problemes que es comenten a ( annex2 ), el primer que vam fer és anar a la pàgina (UCSC Genome Browser on Human July 2003 Freeze).Per a més informació passar a l'apartat de Resultats .
Vam mirar si les anotacions d'inici i final de transcripció dels 23 gens trobats coincidien amb algun de la base de dades de RefSeq i Ensembl, si era així ja no continuaríem treballant amb aquestes anotacions, ara bé, si aquests gens només els trobàvem a geneid i genscan o un dels dos, seria amb aquestes anotacions amb les que faríem la futura predicció de gens.
D'aquesta manera vam passar de 23 gens a 7 que es mostren a la taula següent:
| CROMOSOMA | IDENTIFICADOR |
| chr2 | chr2_2065.1 |
|---|---|
| chr5 | chr5_1143.1 |
| chr6 | chr6_928.1 |
| chr6_1127.1 | |
| chr7 | chr7_292.1 |
| chr7_1204.1 | |
| chr7_1259.1 |
D'aquests 7 possibles gens vam fer un megablast sobre la base de dades de ESTs humans. Per veure mes informacio anar a Resultats . Els resultats del megablast els vam col.locar en els següents fitxers:
blastchr2_2065.1.txt
blastchr5_1143.1.txt
blastchr6_928.1.txt
blastchr6_1127.1.txt
blastchr7_292.1.txt
blastchr7_1204.1.txt
blastchr7_1259.1.txt
Ara aquests fitxers els modificarem per tal d'aconseguir les anotacions dels ESTs:
Primer farem:bash$ grep -v "#" blastchr6_928.1.txt | gawk '{print $2}' | uniq -c > genchr6_928.1.txt
genchr2_2065.1.txt
genchr5_1143.1.txt
genchr6_928.1.txt
genchr6_1127.1.txt
genchr7_292.1.txt
genchr7_1204.1.txt
genchr7_1259.1.txt On obtindrem les anotacions de tots el ESTs de la sortida del blast pels diferents gens; a més també tindrem les repeticions de cadascun d'aquest ESTs, que seran important ja que nosaltres només ens quedarem amb els que surten més d'un cop. Ja que la presencia de més d'un EST a una certa regió del gen, reforça més la hipòtesi que aquest gen pugui ser real, no en canvi, la presencia d'un unic EST. Per tant amb la próxima comanda farem una llista amb la qual tindrem només l'anotació dels ESTs repetits més d'un cop:
bash$ gawk '{if ($1>1){print}}'genchr2_2065.1.txt | perl -ne '/(\w+)\Z/;print "$1\n"'> achr2_2065.txt Tenint:
achr2_2065.1.txt
achr5_1143.1.txt
achr6_928.1.txt
achr6_1127.1.txt
achr7_292.1.txt
achr7_1204.1.txt En aquest cas, no tenim fitxer de l'anotació chr7_1259.1 perquè ara només hi ha un ESTs diferent que creua un intró, i com això ho prenem com a dolent ja que no reforça la hipòtesis, ja no treballarem amb aquest fitxer.
El pas següent és aconseguir les seqüències fasta dels ESTs dels diferents fitxers achr_.txt. Per a fer-ho, vam anar a la pàgina del NCBI --> Entrez-->nucleotide--> entrez tools-->Batch Entrez (ncbi)
3.Visualització dels resultats
.Ara hem d'aconseguir visualitzar amb un plot els resultats, és a dir, veure si hi ha ESTs que corroborin l'existència d'algun gen real:
chr2_2065.1.ests.fa
chr5_1143.1.ests.fa
chr6_928.1.ests.fa
chr6_1127.1.ests.fa
chr7_292.1.ests.fa
chr7_1204.1.ests.fa
Pels ESTs:
- bash$ sed 's/|/_/g;s/ /_/g' chr6_1127.ests.fa | gawk '{if ($1~/>/){print substr($1,1,30)}else{print $0}}' > chr6_1127.ests2.fa
Per tal que la primera línia (anotació del ESTs quedi més reduïda i no tinguem problemes alhora de continuar amb el treball).
- bash$ est2genome -est chr6_1127.ests2.fa -genome ../chr6_1127.1.fa > chr6_11127.estaln.txt
- bash$ est2genome2gff.pl 0 100000 chr6_1127.estaln.txt > chr6_1127.estaln.gff
Amb això aconseguim els fitxers en format gff.
Ara bé, en la resta dels casos el strand del gen és negatiu, per tant, hem d'aplicar la següent comanda:Pel GEN:
- grep chr6_1127 ../final.txt | refgene2gff.pl chr6_1127 | gawk 'BEGIN{OFS="\t"}{$2=$1;$4+=1000;$5+=1000;print $0}' | grep -v Intron > chr6_1127.gen.gff
Cal tenir en compte el 1000 parells de bases que havíem afegit, per això fem $4+1000 i $5+1000
- bash$ gawk'{if ($1~/>/){printf "%s\t",$0}else{printf "%s",$1}}' chr6_1127.1.fa | perl -ne '/(\S+)\s+(\w+)/;$i=$1;$s=$2;$s=~tr/ACGTacgt/TGCAtgca/;$s=reverse($s);printf "%s\n%s",$i,$s' | fold -60 > chr6_1127.1.genenforward.fa
chr5_1143.1.gen.genenforward.fa
chr6_928.1.gen.genenforward.fa
chr6_1127.1.gen.genenforward.fa
chr7_292.1.gen.genenforward.fa
chr7_1204.1.gen.genenforward.fa
- bash$ grep "chr6_1127" final.txt | refgene2gff.pl -f chr6_1127 | gawk 'BEGIN{OFS="\t"}{$2=$1;$4+=1000;$5+=1000;print $0}' | grep -v Intron > chr6_1127.gen.genenforward.gff
chr5_1143.1.gen.genenforward.gff
chr6_928.1.gen.genenforward.gff
chr6_1127.1.gen.genenforward.gff
chr7_292.1.gen.genenforward.gff
chr7_1204.1.gen.genenforward.gff
Cal tenir en compte el 1000 parells de bases que havíem afegit, per això fem $4+=1000 i $5+=1000
Ara, ja per finalitzar, es tracte de visualitzar aquests resultats, per tant:
- bash$ gff2ps chr6_1127.gen.gff chr6_1127.estaln.gff > chr6_1127.ps
o bé, aquesta altra si l'anotació està pels forward:
- bash$ gff2ps -r chr6_1127.gen.genenforward.gff chr6_1127.estaln.gff > chr6_1127.ps
- bash$ convert -rotate 90 -geometry 600x400 chr6_1127.ps chr6__1127.jpg i ja per acabar visualtzar els resultats amb kviev:
- kview chr6_1127.jpg
|
|