La seqüència AC010727 està formada per un total de 148061 parells de bases. Aquí es poden veure les freqüècies absolutes i relatives, respectivament, d'aparició de cada un dels nucleòtids (han estat obtingudes per una serie de comandes en awk):
Adenina 45202 0.3053 Citosina 33108 0.2256 Guanina 29905 0.2020 Timidina 39852 0.2692Com es pot observar el contingut en A i T és una mica superior al de C i G, igual que en el conjunt del genoma humà.
Identificació dels elements de repetició
Degut al fet que només una petita part del genoma és regió codificant i que molt bona part està formada per regions repetitives, per poder fer un estudi de les regions gèniques de la seqüència és interessant localitzar en primer lloc els element repetitius.
S'ha enviat la seqüència en format fasta al servidor del EMBL RepeatMasker. Aquest programa d'identificació de regions repetitives retorna els resultats en forma de tres fitxers.
En el primer fitxer de sortida, repmask.out, hi ha una llista amb tots els elements repetitius ordenats per ordre d'aparició. També hi ha la informació sobre el tipus d'element repetitiu trobat, la seva localització concreta i el seu score (entre altres paràmetres). Aquest fitxer de sortida és el que s'ha utilitzat per veure les localitzacions del elements repetitius en la seqüència de forma gràfica. Per obtenir la imatge s'ha reorganitzat l'arxiu per tenir-lo en format gff (per comandes awk), i després s'ha introduït en el programa gff2ps que prèviament s'ha instal·lat localment.

FIG 1. Alineament de les seqüències repetitives.
És molt difícil interpretar la informació obtinguda a partir del fitxer anterior tot i que gràficament podem observar que hi ha una zona entre 15000 i 60000 parells de bases on hi ha acumulació d'elements ALU (correpon a la línia 6 a partir del final). El fet que en el genoma humà es correlacioni abundància d'elements ALUs i regions codificants suggereix que podria haver-hi una regió exònica en aquest segment de la seqüència.
El segon fitxer obtingut pel RepeatMasker és una recopilació de la informació en forma de taula:
number of elements* length occupied percentagef of sequence
SINEs: 69 14253 bp 9.63 %
ALUs 35 8971 bp 6.06 %
MIRs 34 5282 bp 3.57 %
LINEs: 52 16836 bp 11.37%
LINE1 23 9737 bp 6.58 %
LINE2 29 7099 bp 4.79 %
L3/CR1 0 0 bp 0.00 %
LTR elements: 23 9189 bp 6.21 %
MaLRs 14 6525 bp 4.41 %
ERVL 5 1652 bp 1.12 %
ERV_classI 2 601 bp 0.41 %
ERV_classII 0 0 bp 0.00 %
DNA elements: 33 7699 bp 5.20 %
MER1_type 27 5845 bp 3.95 %
MER2_type 3 1600 bp 1.08 %
Unclassified: 0 0 bp 0.00 %
Total interspersed repeats: 47977 bp 32.40%
Small RNA: 0 0 bp 0.00 %
Satellites: 0 0 bp 0.00 %
Simple repeats: 29 2211 bp 1.49 %
Low complexity: 19 836 bp 0.56 %
* most repeats fragmented by insertions or deletions have been counted as one element
S'ha detectat que els elements LINEs, SINEs i LTR representen aproximadament el 32,40% de la seqüència problema, valor inferior al 45% que s'observen en el conjunt del genoma. Entre aquests tipus d'elements repetitius podem destacar un contingut de 6.06% d'elements ALUs, que com s'ha comentat anteriorment estan principalment agrupats en una regió.
Les regions codificants també es caracteritzen per un contingut més elevat en G i C respecte a regions no codificants. El document obtingut del Repeatmasker també mostra el contingut en G i C de la seqüència i correspon al 42.55 %. Aquest fet no contradiu la possibilitat que aquesta seqüència tan llarga pogui contenir gens.
Finalment el tercer arxiu obtingut és la seqüència emmascarada en format fasta que posteriorment serà utilitzada en programes de predicció de gens.
Geneid
El primer que s'ha fet és instal.lar el programa
geneid localment per tal d'obtenir els resultats de forma més ràpida.
S'ha fet córrer la seqüència emmascarada i sense emmascarar en format FASTA.
Els resultats han estat obtinguts en format gff per la seqüència emmascarada i la no emmascarada. Aquest format permet visualitzar les prediccions a través del programa ghostview gràcies a la transformació amb el programa gff2ps. A més, també s'han extret els resultats en format txt que permet visualitzar, entre altres coses, la seqüència d'aminoàcids predita.
Genscan
Per córrer la seqüència en aquest programa s'ha accedit al servidor del genscan i s'ha fet un "paste" en format FASTA deixant els paràmetres que es seleccionen per defecte.
Els resultats s'han obtingut en format txt.
Per tal de visualitzar les prediccions obtingudes amb el programa ghostview s'han reformatejat els fitxers txt de la seqüència (emmascarada i sense emmascarar) en format gff amb un seguit de comandes en llenguatge awk.
MetaGene
El MetaGene es un servidor que et permet córrer diferents programes de predicció de gens a la vegada. Un cop accedit al servidor del MetaGene es fa un "paste" de la seqüència a estudiar (també en format FASTA) i s'ecullen els programes de predicció de gens que es volen utilitzar. En aquest estudi s'han utilitzat tots els programes excepte els ja esmentats en paràgrafs anteriors (geneid i genscan).
Només s'han obtingut resultats a través dels programes Grail i GenMark (en format txt). Aquests programes no han predit estructures gèniques, sinó que només han predit possibles regions codificants sense relacionar-les entre elles. Per aquest motiu, no s'ha cregut convenient d'estudiar la seqüència en la seva forma emmascarada i sense emmascarar, sinó que només s'ha estudiat la seqüència sense tenir en compte totes les regions repetitives de DNA. Per tant, només s'ha reformatejat a gff la sortida dels programes Grail i GeneMark de la seqüència emmascarada.
Un cop recollides totes les sortides dels programes de predicció de gens, s'analitza si els diferents programes localitzen les estructures gèniques en les mateixes posicions de la seqüència estudiada.
Per desenvolupar aquest pas s'han necessitat els següents programes:
-gff2ps (programa que permet passar de format gff a format postcript -ps-)
-Ghostview (programa que permet visualitzar com s'aparellen diferents regions de DNA amb la seqüència a estudiar a partir de fitxers en format ps)
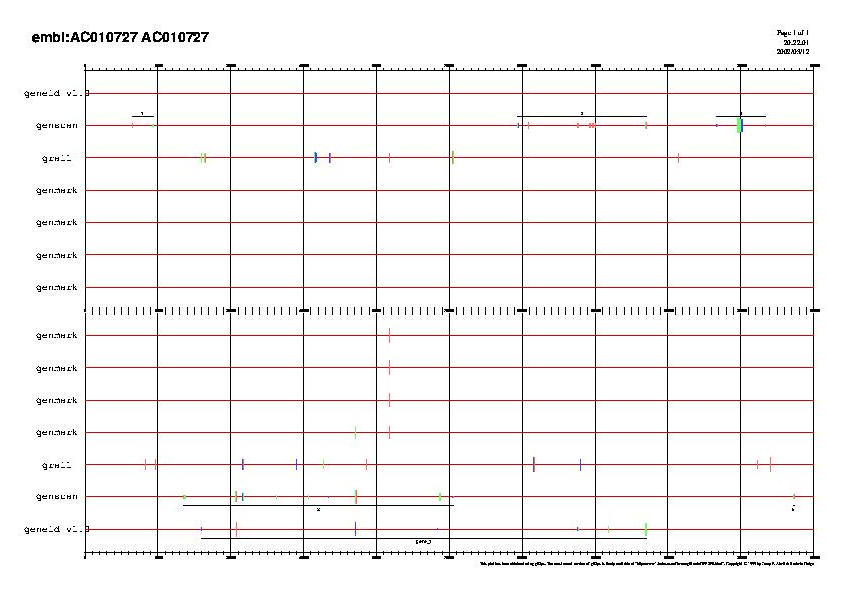
FIG 2. Comparació dels resultats obtinguts a partir dels programes geneid, genscan, Grail, GeneMark per la seqüència emmascarada.
Al comparar les prediccions obtingudes amb el geneid, genscan, Grail i GeneMark s'observa que els diferents programes no coincideixen a l'hora de predir exons. El Grail i el GeneMark prediuen regions codificants en zones que no es suporten en cap dels altres programes de predicció de gens. En canvi, es detecte que tant el geneid com el genscan comparteixen algunes de les prediccions. Per fer una millor comparació d'aquestes regions codificants compartides s'ha fet una visualització excloent els programes Grail i GeneMark que no reforçen cap predicció.
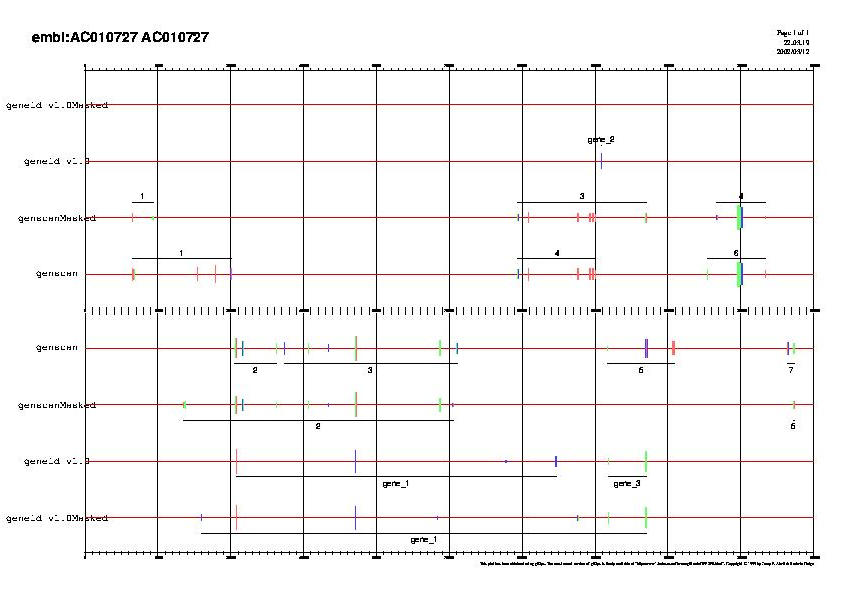
FIG 3. Comparació dels resultats obtinguts a partir dels programes geneid i genscan per la seqüència emmascarada i sense emmascarar.
El programa genscan prediu estructures exòniques en sentit directe i revers.
En sentit directe prediu 3 gens tant en la seqüència emmascarada com en la no emmascarada, però l'estructura exònica d'aquests gens predits en tots dos casos no segueix exactament el mateix patró.
En sentit revers s'observen més diferències en funció de si s'analitza la seqüència emmascarada o sense emmascarar. En el primer cas, es prediuen 2 gens: el primer està format per 9 exons (3 dels quals tenen bastanta força i també són predits en la seqüència sense emmascarar) i l'altre només conté un exó de poca força. Mentre que en el segon cas, es perdiuen 4 gens diferents.
El programa geneid prediu estructures exòniques en sentit directe i revers, tot i que en sentit directe només detecta un exó de poca força al analitzar la seqüència no emmascarada.
En sentit revers les prediccions fetes en la seqüència emmascarada i no emmascarada són pràcticament equivalents, tot i que en cada cas els mateixos exons estiguin agrupats en gens diferents.
Al analitzar el gràfic de forma global s'observa que hi ha 3 possibles regions codificants en sentit revers que surten en la majoria de prediccions. Aquest fet suggereix l'existència de 3 exons que podrien formar part d'un gen. En canvi, en sentit directe no s'observa cap estructura codificant reforçada per tots els programes, per això s'assumeix que és poc probable de trobar un gen en aquest sentit.
Amb els programes de predicció de gens utilitzats, es pot formular una primera hipòtesi d'on es localitzen les regions codificants contingudes en la seqüència AC010727. Aquesta hipòtesi prediu que la regió genòmica estudiada conté un gen en sentit revers format per 3 exons.
Degut a que la predicció del programa geneid amb la seqüència emmascarada és la que s'aproxima més a aquesta primera hipòtesi formulada, a partir d'ara es treballarà amb la la seqüència AC10727 emmascarada, i l'estudi es basarà amb els resultats obtinguts a partir del programa geneid.
Validació de la predicció de gens
El següent pas és validar el patró exònic trobat fins al moment i intentar identificar possibles exons no reconeguts amb els programes de predicció de gens utilitzats.
Aquest pas es desenvolupa amb l'ajuda del MEGABLAST que es tracte d'un programa que aparella totes les possibles seqüències emmagatzemades en la Base de Dades seleccionada amb la seqüència problema.
S'accedeix al servidor ncbi blast i es selecciona la opció "MEGABLAST" on es fa un "paste" de la seqüència estudiada amb el paràmetre base de dades de EST humans.
El resultat obtingut es tot un llistat dels EST (expressed sequence tag) humans continguts en la Base de Dades seleccionada que s'aparellen amb la nostra seqüència.
Els EST són petites seqüències de RNAm que han estat seqüenciades un cop s'han passat a cDNA per retrotranscripció. Per tant, aquests alineaments aporten informació de possibles regions codificants.
Si els EST trobats pel programa Megablast s'aparellen en aquelles regions de la seqüència estudiada on s'han predit els 3 exons, s'obtindrà un reforçament de la hipòtesi formulada.
Per analitzar els aparellaments obtinguts es visualitza amb el programa ghostview que només pot llegir fitxers en format ps. Per aquest motiu es reformateja la sortida del programa Metablast en gff. Aquesta transformació es pot fer amb un programa en perl com parseblast programat per Josep Abril. Amb el fitxer en gff es passa a format ps a través del programa gff2ps.
Es pot veure que hi ha un gran nombre d'ESTs que s'aparellen amb la seqüència estudiada. La major part d'aquests s'han aparellat en forma d'splicing, és a dir que diferents regions del EST s'han aparellat en diferents regions de la seqüència. Però també es pot veure un gran nombre d'ESTs dispersats al llarg de la seqüència de forma aïllada, possiblement es tracte d'artefactes que es produeixen durant la construcció de les biblioteques d'ESTs. Per aquest motiu i pel fet que, en general, només es consideren bones evidències de l'existčncia de gens els ESTs que provenen de RNAtp que han passat per splicing, aquest anàlisi es centrerà en els spliced ESTs.
Un altre factor que s'ha de tenir en compte quan es treballa amb ESTs és si es tracta de EST 3 prima o EST 5 prima. La majoria dels ESTs obtinguts estan anotats a la base de dades com a 3 prima. En canvi, aquells ESTs que s'alinien en 4 regions són 5 prima. El problema és que en una mateixa regió
s'hi han aparellat tan ESTs 5 prima com ESTs 3 prima. A més, ESTs que coincideixen en el patró d'aparellament tenen anotacions contràries. Per aquestes raons es sospita que hi ha un mal anotament dels EST. Per comprovar que es tracta d'un error s'ha fet un alineament entre dos ESTs que coincideixen en patró i divergeixen en anotació amb el CLUSTALW. Com era d'esperar s'ha obtingut un alineament amb un score elevat (98% d'alineament). Per aquest fet en la resta de l'estudi no es tindrà en compte les anotacions dels ESTs. Aquests errors poden ser causats pel fet que el RNA estigués degradat en el moment que es va començar a seqüenciar per 3 prima.
Per tal de desfer-nos dels ESTs que no són interessants per l'anàlisi, s'aplica al fitxer gff un programa per eliminar tots aquells que només s'aparellen una vegada. A més a més, s'ha introduït el sentit de l'aparellament. Al visualitzar aquests resultats (document no mostrat) es destaca que la gran majoria dels EST s'aparellen amb la seqüència reverse, per això també s'ha decidit elimiar del fitxer gff els EST forward.
De tots els EST que queden deprés d'aquesta primera selecció es destaca que hi ha 8 patrons d'aparellament diferents. S'ha agafat de forma aleatòria un representant de cada patró per poder tenir una millor visualització com es pot veure a la figura 4.
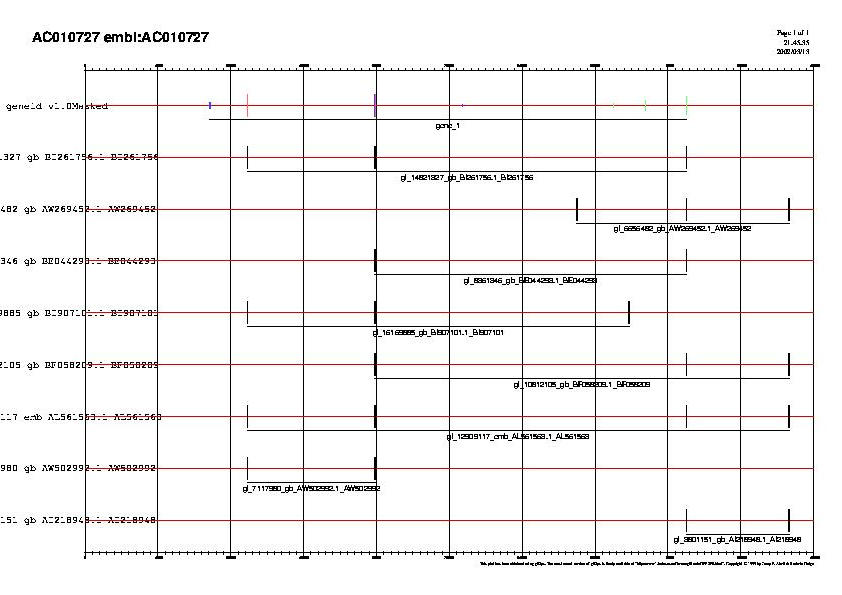
FIG 4. Visualització dels 8 patrons d'ESTs i de les prediccions del geneid (reverse strand).
La major part dels EST es troben agrupats en 4 grans regions (dades corresponents al EST AL561563):
-31133 a 31244
-55612 a 55798
-115486 a 115632
-135197 a 135410
Les 3 primeres s'aparellen a les mateixes posicions que els 3 exons de la Hipòtesi formulada reforçant-la. Però a més a més es pot obsevar que la quarta regió està molt suportada pel conjunt dels EST i que es correspon amb una regió predita pel genscan amb la seqüència no emmascarada. Per aquesta raó s'ha decidit de buscar, a la base de dades del ncbi, les seqüències dels EST per fer un aparellament de manera més acurada amb el programa est2genome. Els resultats obtinguts han estat reveladors, ja que patrons de només dos aparellaments han trobat noves regions d'homologia. D'aquesta manera alguns del 8 patrons seleccionats anteriorment han desaparegut per convertir-se en algun dels altres. A més d'aquests 8 ESTs se n'han analitzat més per assegurar que no ha estat una quüestió d'atzar, i s'han repetit els mateixos resultats.
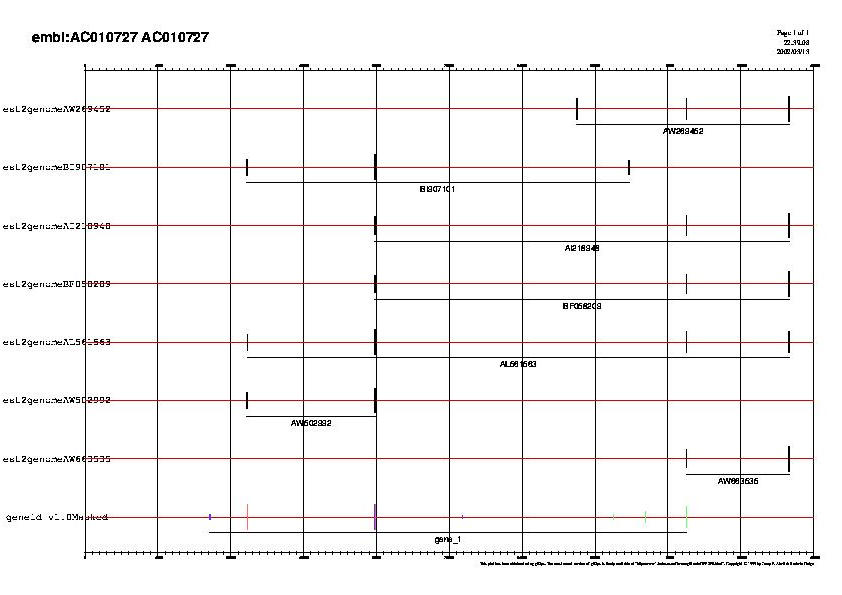
FIG 5. Visualització dels patrons d'ESTs obtinguts amb el est2genome i de les prediccions del geneid (reverse strand) .
En aquest moment s'ha decidit de tornar a passar la seqüència problema al programa geneid introduïnt evidències d'homologia en la regió que aquest programa no havia predit cap estructura exònica amb un score suficientment significatiu. Aquest pas es fa per veure si, d'aquesta manera, el geneid prediu un gen que englobi les 4 regions codificants evidenciades pels ESTs.
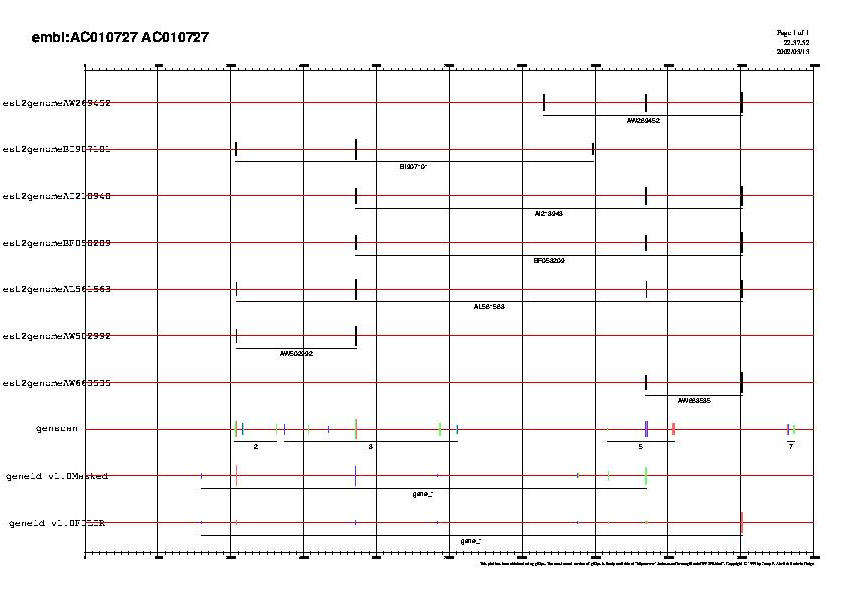
FIG 6. Visualització del geneid amb evidències de similaritat, el genscan sense emmascarar i els aparellaments dels EST obtinguts amb el est2genome.
Tal com es volia el geneid ha predit les quatre regions codificants, però es pot observar que continua predint dos exons més.
També es pot veure que el fet d'afegir aquesta evidència ha provocat una disminució important de la força de tots els altres exons.
A partir de tots els resultats del programes de predicció de gens i els reforçaments dels ESTs es pot formular la hipòtesi de quatre regions codificants que formarien part d'un sol gen (sense confirmació). En canvi es desconeix si aquest gen predit està format exclusivament per aquests quatre exons, o es tracta d'un gen més llarg que está clonat en diferents BACs contigs, de manera que la seqüència estudiada només contindria el principi, el final, o un fragment intern del gen.
Estudi de la regió reguladora dels gens predits
A partir dels resultats anteriors no es pot dir que el primer exó obtingut en la regió 5 prima de la seqüència reverse sigui un start, a més a més aquest és l'exó que està menys sostingut per les prediccions i un cop corregut el geneid amb evidències tampoc apareix com a primer exó.
S'ha fet un blastp amb la seqüència d'aminoàcids obtinguda a partir de les prediccions del geneid. El resultat obtingut és l'existència d'una proteïna hipotètica anomenada KIAA0084. En aquesta pàgina també hi ha indicat el número de identificació Q14699 per poder accedir a la informació de la base de dades Swissprot. A partit d'aquesta s'ha arribat a l'Ensembl on s'ha trobat informació sobre els exons predits.
Aquesta proteïna hipotètica estaria formada per un total de 10 exons distribuïts en diferents BACs. Justament els 4 primers exons corresponen al BAC de identificació AC010727 que és el subjecte de l'anàlisi, coincidint amb una de les possibles hipòtesis. De manera que el primer exó seria un iniciador. Al comparar les coordenades de les prediccions de la base de dades i del geneid es veu que difereixen molt poc:
Ensembl geneid
--------------------------------------------------------------------------
Inici Final Inici Final
--------------------------------------------------------------------------
exo 1 135200 135401 135200 135388
exo 2 115483 115635 115483 115697
exo 3 55609 55795 55609 55736
exo 4 31133 31241 31133 31241
Aquests resultats suggereixen que l'inici de la traducció del gen, si existeix, pot estar al voltant del nucleòtid 135400. Com que no es coneix exactament l'inici de la transcripció, per esbrinar si la regió upstream podria ser una regió reguladora, s'han agafat uns 2000 parells de bases per sobre per fer-ne un anàlisi.
Per poder extreure fragments parcials a partir de la seqüència completa s'ha creat el programa pickaseq en llenguatge perl. Per executar-lo s'ha d'introduir en primer lloc el fitxer d'on es vol extreure la seqüència, seguit del primer nucleòtid que es vol agafar i finalment la llargada desitjada. Així s'ha obtingut un fitxer amb 2012 pb upstream del nucleòtid 135400.
En primer lloc s'ha enviat aquesta seqüència al programa TRANSFAC que cerca possibles llocs d'unió de factors de transcripció en la seqüència enviada. Inicialment s'han buscat llocs d'unió amb totes les matrius de vertebrats. S'han modificat els paràmetres per obtenir un menor número de resultats però de més similaritat: core similarity = 1.00 i matrix similarity 0.90. Els resultats obtinguts han estat transformats a format gff per poder visualitzar-los amb ghostview un cop transformats pel gff2ps.
En segon lloc s'ha seleccionat de forma exclusiva la matriu V$TATA_01 per detectar si a la regió estudiada hi ha alguna TATA box (core similarity = 1.00 i matrix similarity 0.95). Els resultats obtinguts han estat agrupats en la figura 7 amb els possibles llocs d'unió dels factors de transcripció.
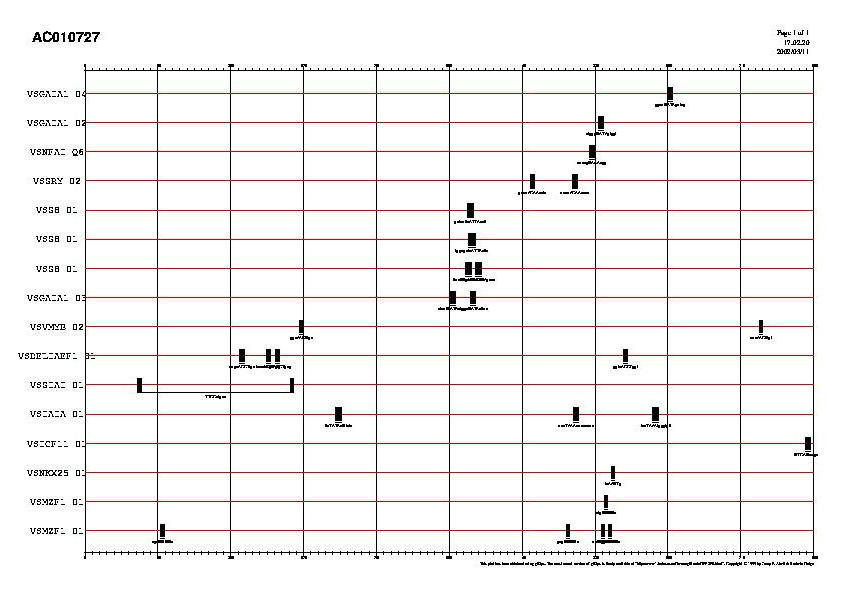
Fig 7. Possibles sites d'unió de factors de transcripció i TATA box en 2000 nucleòtids upstream.
El programa detecta tres possibles TATA box (5Ş fila a partir del final del document), una a partir del nucleòtid 136063, una altra al 136683 i la última al 136891 respecte la numeració inicial de la seqüència. També es pot observar que al voltant d'aquestes zones hi ha regions d'unió de diversos factors de transcripció.
Aquests resultats reforcen la hipòtesi que aquest primer exó sigui un start.
Posteriorment s'ha fet un estudi de possibles regions reguladores en els introns. Per això s'ha fragmentat la seqüència inicial amb el programa pickaseq en: intró 1, intró 2, intró 3, intró 4; també s'han obtingut els exons: exó 1, exó 2, exó 3, exó 4.
S'han enviat de forma seqüencial cada un dels introns al programa TRANSFAC, en aquests cas s'han buscat en matrius de vertebrats però els paràmetres han estat fixats en core similarity = 1.00 i matrix similarity 0.99, ja que degut a la llargada de la seqüencia intrònica amb valors inferiors s'obtenen molts més resultats. Tots els resultats obtinguts s'han agrupat en un mateix fitxer transformat a format gff. Finalment s'ha obtingut la imatge amb el programa gff2ps.
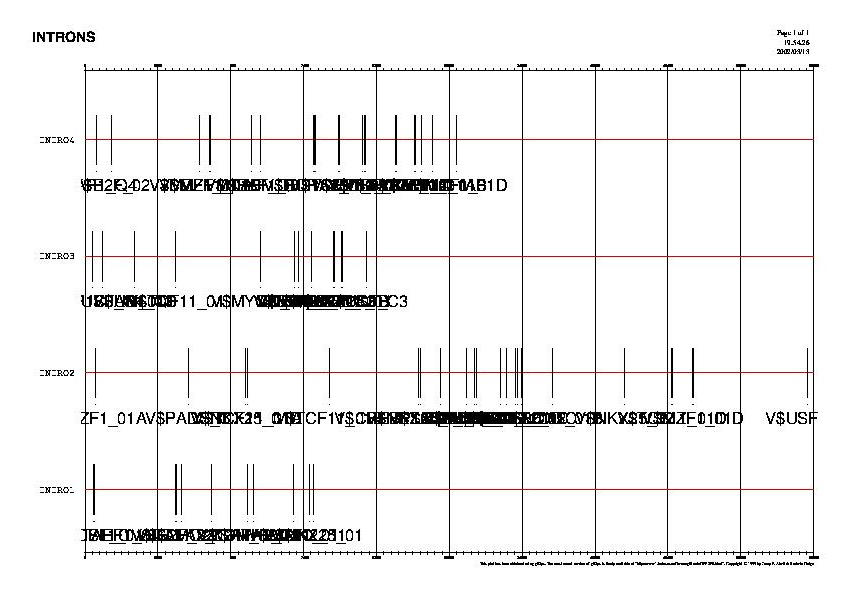
Fig 8. Llocs d'unió de factors de transcripció a cada un dels introns.
Aquests resultats mostren que a les regions intròniques hi ha seqüències de DNA susceptibles de ser reconegudes per factors de transcripció. Aquest fet suggereix l'exitència d'una possible regulació a nivell intrònic. Però els resultats obtinguts no permeten confirmar aquesta possibilitat.
Per acabar, s'ha fet un càlcul de la raó de versemblaça a partir de cada un dels fragments intrònics o exònics obtinguts, amb l'ajuda d'un programa que s'havia utilitzat a pràctiques. Els resultats estan recopilats a la següent taula:
exo 1 exo 2 exo 3 exo 4 intro 1 intro 2 intro 3 intro 4
------------------------------------------------------------------------------------------------------------------------------------------
pauta 1 -3.21 -5.29 -8.76 -80.58 -9000 -28043 -11027 -13433
pauta 2 -5.93 -240 9.32 -84.46 -8384 -28552 -11175 -12804
pauta 3 -3.87 -79.53 2.79 -0.18 -9266 -28727 -11635 -14098
Aquests resultats no són els esperats. En gens codificants s'esperaria obtenir valors positius en els exons (al menys en la pauta de lectura correcta), i valors negatius a la resta d'exons i als introns. Només s'obtenen valors positius per dues pautes de lectura de l'exó 3. Això indica que és més probable trobar aquesta seqüència en una regió codificant que en regions no codificants. En tots els altres casos on s'observen valors negatius es pot veure que la raó de versemblaça dels exons és propera a zero i molt superior a la dels introns. Tot i haver obtingut aquests resultats no es pot descartar la hipòtesi formulada.
Conclusions
L'objecte d'aquest projecte és estudiar la seqüència AC010727 localitzada al cromosoma 3.
A partir de la seqüència emmascarada, s'han utilitzat diferents programes de predicció de gens (geneid, genscan, MetaGene). Els resultats obtinguts han estat desiguals. Tot i així, tant les previsions del geneid com les del genescan coincideixen en la predicció de tres regions codificants en sentit revers.
Al córrer la seqüència al MEGABLAST amb la base de dades d'ESTs humans, s'ha reforçat aquesta hipòtesi. Contra tot pronòstic, molts dels ESTs s'han alineat amb una quarta regió només suportada per una de les prediccions (genscan), per la qual cosa no s'havia tingut en compte al formular la hipòtesi inicial. Per obtenir un alineament més acurat, s'han aparellat un a un els diferents ESTs amb la seqüència utilitzant el programa est2genome. A partir d'aquí s'ha formulat una nova hipòtesi que prediu quatre exons en revers.
Per aquesta raó s'ha tornat a córrer el geneid amb evidències d'homologia en aquesta nova regió i ha predit un gen que conté els quatre exons de la hipòtesi.
Amb la seqüència d'aminoàcids predita amb el geneid s'han buscat possibles proteïnes homòlogues (blastp). S'ha trobat l'existència d'una proteïna hipotètica (KIAA0084) formada per deu exons dels quals els quatre primers exons es corresponen amb la hipòtesi formula anteriorment. Per aquesta raó s'ha considerat que el primer exó és un start i s'ha fet un estudi agafant 2000 pb usptream per analitzar possibles regions reguladores. A més s'han estudiat individualment possibles regulacions a nivell intrònic.
En base a aquest resultats es pot formular la hipòtesi final:
la seqüència AC010727 conté en sentit revers el fragment de la regió inicial d'un gen. Concretament s'ha trobat una possible regió de regulació on es destaquen tres possibles TATA box i quatre exons.
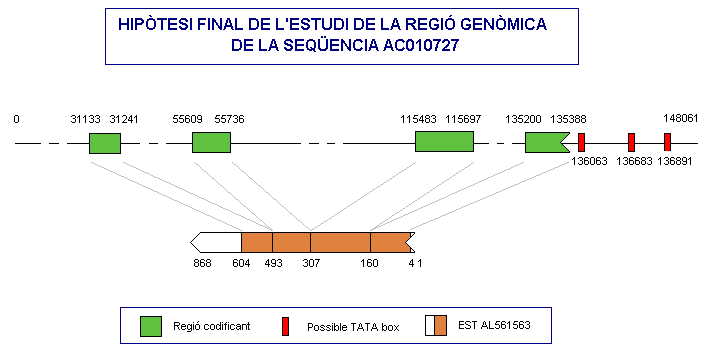
FIG 9.Hipòtesi final de l'estudi de la regió genòmica AC010727.
Autors i agraïments
Els autors d'aquest document són Gina Abelló Sumpsi i Roser Corominas Castiñeira, alumnes de quart de Biologia a la Facultat de Ciències de la Salut i de la Vida de la Universitat Pompeu Fabra.
Adreces de correu electrònic:
gina.abello01@campus.upf.edu
roser.corominas01@campus.upf.edu
Aquest és el treball que han realitzat per l'assignatura de Bioinformàtica cursada l'any 2002.
Finalment s'agraeix als membres del Grup de Recerca en Informŕtica Biomčdica de la UPF i IMIM la seva ajuda i pacičncia.
Software i bibliografia electrònica
SERVIDORS:
1. RepeatMasker http://woody.embl-heidelberg.de/repeatmask/
2. gff2ps http://genome.imim.es/software/gfftools/GFF2PS.html
3. geneid http://genome.imim.es/software/geneid/index.html
4. genescan http://genes.mit.edu/GENSCAN.html
5. MetaGene http://rgd.mcw.edu/METAGENE/
6. Grail http://compbio.ornl.gov/Grail-1.3/
7. GenMark http://dixie.biology.gatech.edu/GeneMark/genemark24.cgi
8. ncbi http://www.ncbi.nlm.nih.gov/blast/. En aquest servidor s'han utilitzat els progrmanes Standard protein-protein BLAST [blastp] i MEGABLAST.
9. est2genome http://bioweb.pasteur.fr/seqanal/interfaces/est2genome.html
10. Swissprot http://www.expasy.ch/sprot/
11. Ensembl http://www.ensembl.org/Homo_sapiens/geneview?gene=ENSG00000131378
12. TRANSFAC http://transfac.gbf.de/cgi-bin/matSearch/matsearch.pl/
13. CLUSTALW http://www2.ebi.ac.uk/clustalw
PROGRAMES:
14. ghostview
15. parseblast
16. pickaseq
17. dnaloglkh
S'ha fet un blastp amb la seqüència d'aminoàcids obtinguda a partir de les prediccions del geneid. El resultat obtingut és l'existència d'una proteïna hipotètica anomenada KIAA0084. En aquesta pàgina també hi ha indicat el número de identificació Q14699 per poder accedir a la informació de la base de dades Swissprot. A partit d'aquesta s'ha arribat a l'Ensembl on s'ha trobat informació sobre els exons predits.
Aquesta proteïna hipotètica estaria formada per un total de 10 exons distribuïts en diferents BACs. Justament els 4 primers exons corresponen al BAC de identificació AC010727 que és el subjecte de l'anàlisi, coincidint amb una de les possibles hipòtesis. De manera que el primer exó seria un iniciador. Al comparar les coordenades de les prediccions de la base de dades i del geneid es veu que difereixen molt poc:
Ensembl geneid -------------------------------------------------------------------------- Inici Final Inici Final -------------------------------------------------------------------------- exo 1 135200 135401 135200 135388 exo 2 115483 115635 115483 115697 exo 3 55609 55795 55609 55736 exo 4 31133 31241 31133 31241
A partir de la seqüència emmascarada, s'han utilitzat diferents programes de predicció de gens (geneid, genscan, MetaGene). Els resultats obtinguts han estat desiguals. Tot i així, tant les previsions del geneid com les del genescan coincideixen en la predicció de tres regions codificants en sentit revers.
Al córrer la seqüència al MEGABLAST amb la base de dades d'ESTs humans, s'ha reforçat aquesta hipòtesi. Contra tot pronòstic, molts dels ESTs s'han alineat amb una quarta regió només suportada per una de les prediccions (genscan), per la qual cosa no s'havia tingut en compte al formular la hipòtesi inicial. Per obtenir un alineament més acurat, s'han aparellat un a un els diferents ESTs amb la seqüència utilitzant el programa est2genome. A partir d'aquí s'ha formulat una nova hipòtesi que prediu quatre exons en revers. Per aquesta raó s'ha tornat a córrer el geneid amb evidències d'homologia en aquesta nova regió i ha predit un gen que conté els quatre exons de la hipòtesi.
Amb la seqüència d'aminoàcids predita amb el geneid s'han buscat possibles proteïnes homòlogues (blastp). S'ha trobat l'existència d'una proteïna hipotètica (KIAA0084) formada per deu exons dels quals els quatre primers exons es corresponen amb la hipòtesi formula anteriorment. Per aquesta raó s'ha considerat que el primer exó és un start i s'ha fet un estudi agafant 2000 pb usptream per analitzar possibles regions reguladores. A més s'han estudiat individualment possibles regulacions a nivell intrònic.
En base a aquest resultats es pot formular la hipòtesi final:
la seqüència AC010727 conté en sentit revers el fragment de la regió inicial d'un gen. Concretament s'ha trobat una possible regió de regulació on es destaquen tres possibles TATA box i quatre exons.
FIG 9.Hipòtesi final de l'estudi de la regió genòmica AC010727.
Autors i agraïments
Els autors d'aquest document són Gina Abelló Sumpsi i Roser Corominas Castiñeira, alumnes de quart de Biologia a la Facultat de Ciències de la Salut i de la Vida de la Universitat Pompeu Fabra.
Adreces de correu electrònic:
gina.abello01@campus.upf.edu
roser.corominas01@campus.upf.edu
Aquest és el treball que han realitzat per l'assignatura de Bioinformàtica cursada l'any 2002.
Finalment s'agraeix als membres del Grup de Recerca en Informŕtica Biomčdica de la UPF i IMIM la seva ajuda i pacičncia.
Software i bibliografia electrònica
SERVIDORS:
1. RepeatMasker http://woody.embl-heidelberg.de/repeatmask/
2. gff2ps http://genome.imim.es/software/gfftools/GFF2PS.html
3. geneid http://genome.imim.es/software/geneid/index.html
4. genescan http://genes.mit.edu/GENSCAN.html
5. MetaGene http://rgd.mcw.edu/METAGENE/
6. Grail http://compbio.ornl.gov/Grail-1.3/
7. GenMark http://dixie.biology.gatech.edu/GeneMark/genemark24.cgi
8. ncbi http://www.ncbi.nlm.nih.gov/blast/. En aquest servidor s'han utilitzat els progrmanes Standard protein-protein BLAST [blastp] i MEGABLAST.
9. est2genome http://bioweb.pasteur.fr/seqanal/interfaces/est2genome.html
10. Swissprot http://www.expasy.ch/sprot/
11. Ensembl http://www.ensembl.org/Homo_sapiens/geneview?gene=ENSG00000131378
12. TRANSFAC http://transfac.gbf.de/cgi-bin/matSearch/matsearch.pl/
13. CLUSTALW http://www2.ebi.ac.uk/clustalw
PROGRAMES:
14. ghostview
15. parseblast
16. pickaseq
17. dnaloglkh
roser.corominas01@campus.upf.edu
1. RepeatMasker http://woody.embl-heidelberg.de/repeatmask/
2. gff2ps http://genome.imim.es/software/gfftools/GFF2PS.html
3. geneid http://genome.imim.es/software/geneid/index.html
4. genescan http://genes.mit.edu/GENSCAN.html
5. MetaGene http://rgd.mcw.edu/METAGENE/
6. Grail http://compbio.ornl.gov/Grail-1.3/
7. GenMark http://dixie.biology.gatech.edu/GeneMark/genemark24.cgi
8. ncbi http://www.ncbi.nlm.nih.gov/blast/. En aquest servidor s'han utilitzat els progrmanes Standard protein-protein BLAST [blastp] i MEGABLAST.
9. est2genome http://bioweb.pasteur.fr/seqanal/interfaces/est2genome.html
10. Swissprot http://www.expasy.ch/sprot/
11. Ensembl http://www.ensembl.org/Homo_sapiens/geneview?gene=ENSG00000131378
12. TRANSFAC http://transfac.gbf.de/cgi-bin/matSearch/matsearch.pl/
13. CLUSTALW http://www2.ebi.ac.uk/clustalw
PROGRAMES:
14. ghostview
15. parseblast
16. pickaseq
17. dnaloglkh