In search of selenoproteins in the world's smallest mammal
-Craseonycteris thonglongyai-
MATERIALS AND METHODS
The aim of this project is to identify and annotate all the selenoproteins (including its SECIS elements) and the genes that codify for selenoprotein machinery (required for the synthesis of these proteins) found in Craseonycteris thonglongyai. In order to achieve it, we will perform an homology-based study in which we will compare the genome of Craseonycteris thonglongyai with the related proteins described inHomo sapiens.
We have chosen Homo sapiens genome to predict the selenoproteins of Craseonycteris thonglongyai because they are both mammalian animals, so they are quite close in phylogenetic tree. Moreover, it is known that human genome has been exhaustively sequenced and well-annotated. Hence, selenoproteins and genes that codify for selenoproteins machinery are also well annotated.
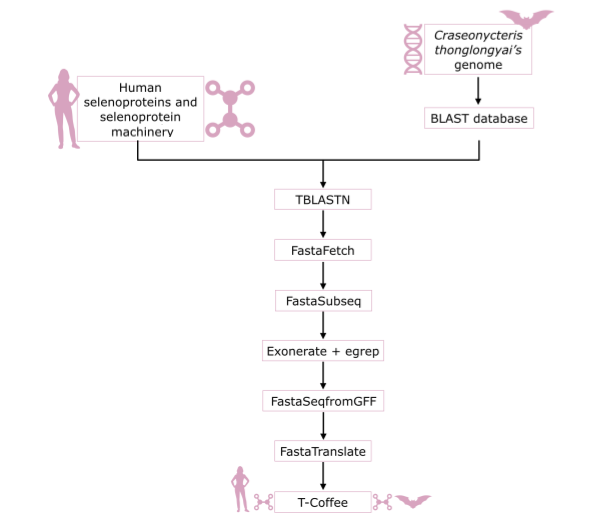
Figure 3. Workflow of the steps performed to predict the selenoproteome of Craseonycteris thonglongyai.
ACQUISITION OF THE GENOME OF Craseonycteris thonglongyai AND THE INDEX:
The genome of Craseonycteris thongongyai was obtained from a directory provided by the teachers of the subject. It could be found by introducing the following path:
As previously mentioned, we decided to compare the genome of Craseonycteris thonglongyai with the human selenoproteome. Therefore, the amino acid sequences of the queries were obtained from SelenoDB 1.0 database.However, some amino acid sequences corresponding to the machinery for the biosynthesis of selenoproteins were download from SelenoDB 2.0 database, because they were not found in SelenoDB 1.0. The amino acid sequences were saved as “name of the protein”.aa.fa.
Finally, since the software does not recognise the “U” character, which corresponds to a Selenocystein in our query sequences, "U" were replaced by an “X” which would be read as “any possible amino acid”. Moreover, all the other symbols that could be found at the end of the sequences such as @, #, etc were also removed for the same reason.
ANNOTATION OF THE SELENOPROTEINS
The first comparisons to predict the potential selenoproteins in Craseonycteris thonglongyai, were performed manually to deeply understand how each step works.
Later, to speed up the process, we designed a semiautomatic perl program. The program automatically runs all the steps needed to predict selenoproteins from the genome of interest by comparison with the query chosen. However, you have to manually acquise the queries and introduce its name in the program to start the comparison. You also have to introduce the name of the scaffold and the first nucleotide aligned. Finally, SECIS prediction was performed with Seblastian, a selenoprotein prediction server (https://seblastian.crg.es/).
To understand better how the semiautomatic perl program works, we will go step by step:
INITIAL STEPS
A folder with all the query sequences in the same directory of the perl file is required to start the program. When you execute the program a list of all the possible queries is printed in the terminal and once you choose your protein of interest, a folder with its name is created.
TBLASTN
After introducing your query of interest, the program automatically runs a TBLASTN which compares the amino acid sequence of the query with the genome of interest. After it, the program asks you in which of the found hits are you interested by letting you choose the percentage of identity and an e-value as a threshold. However, if no alignment can be found, the program will ask you again for a new threshold until it finds at least one alignment that meets the chosen criteria.
The program prints the scaffolds with a hit that meet the criteria chosen before and next, asks you about the quantity of scaffolds of interest. If you are interested in more than one scaffold, the program will analyze independently the ones you are interested in and save the results in different directories inside the directory of the protein. Finally, if you introduce “0” when the program asks for the number of scaffolds to analyze, it will ask you again to choose new criteria to find new scaffolds.
In order to proceed in a similar way with all the proteins, we decided to select scaffolds with significant parameters in the identity and e-value. Hence, the initial value selected for the identity was 90% or greater and for the e-value 0,001 or lower. However, in the cases where no scaffold could be found with such great identity, or where the coverage of the alignment was not greater than the half of the query, we decreased the value in 5 until a scaffold that met the chosen parameters could be found.
Therefore, it can be seen that we focused in the identity more than in the e-value. The reason for this was that most of the scaffolds presented very significant e-values.
It has to be said that in the cases where more than one scaffold appeared with similar identity values, the length of the alignment and its position into the query were considered to decide which one was better. Therefore, if different scaffolds with similar identity and e-value were aligned with the query, but in different positions (in other words, they did not overlapped) all the scaffolds were ran independently, because we hypothesized that the protein was truncated in different scaffolds and so.
Finally, the program saves the file resulting of TBLASTN, called query.blast, in a directory made for that protein.
FASTAFETCH
By introducing the name of the chosen scaffold, fastafetch command allows us to extract the scaffold in which we are interested from the mulifasta genome.fa file by using the index provided by the teachers of the subject. The result will be kept in a file called name of the protein.scaffold.fastafetch.fa in the directory of the selected scaffold.
FASTASUBSEQ
Once the scaffold is extracted from the multifasta file, this command allows us to obtain just the region of interest inside the scaffold where the hit is found. This command will ask us to introduce the position of the first nucleotide in the hit. The start nucleotide of the scaffold in the fastasubseq file will be that one 50.000 positions lower, and the length of the sequence was determined to be 100.000, extending the search in 50.000 nucleotides in both directions from the initial introduced value
Keeping in mind that tblasn only provides hits in exons, we extended by 50.000 nucleotides the extraction so as to avoid the lost of coding sequence because of the presence of introns between the hits. Moreover, in case that nucleotide extension goes beyond scaffold 3’ or 5’ limits, the program will reset the value of the established limits so as to correspond with the start and the end of the scaffold respectively. The extracted sequence will be kept in a file called name of the protein.scaffold.subseq.fa in the directory of the selected scaffold.
EXONERATE
The exonerate command is used to deduce the sequence of the gene from our region of interest. Later, to extract only the exons of the deduced sequence of the gen, the egrep command is used. The obtained sequence will be found in a file called name of the protein.scaffold.exon.gff in the directory of the selected scaffold.
FASTASEQFROMGFF
With the exons extracted from the exonerate prediction, this command deduces the cDNA sequence of the predicted protein. This information will be kept in a file called name of the protein.pred.nuc.fa in the directory of the selected scaffold.
FASTATRANSLATE
This function will translate the cDNA obtained from the previous step into the amino acid sequence. Only the first ORF will be translated since we have used the option 1 in the command -F. After it, the program will check whether in the obtained sequence there is any “U” corresponding to a Selenocystein and will substitute it for a “X” so as to the program can recognise the character. The resulting sequence will be kept in a file called name of the protein.pred.aa.fa in the directory of the selected scaffold.
T-COFFEE
Finally the program will run the T-coffee command which generates a global alignment between the deduced protein sequence from the genome of interest and the initial query from the reference specie. The result is collected in a file called results.name of the protein in the directory of the selected scaffold.
The default values for Gap open penalty and Gap extension penalty were used. The higher the consistency, the better the alignment. The score reported with every T-Coffee alignment is the concistency score, normalized to 1000.
COVERAGE CALCULATION
From the T-coffee alignment, we have calculated two types of coverage. Coverage type I is defined as the percentage of the sequence that has aligned perfectly, only including conserved positions (indicated with *). On the other hand, coverage type II is the percentage of the sequence that has aligned partially, including conserved positions, conservative mutations (:) and semi-conservative mutations (.).
SEBLASTIAN
SECIS elements do not seem to share a similar sequence between each other in order to recognise them. However, it has been shown that they present some kind of estructural conservation. Therefore, they can be identified by studying their structure in 3D. Seblastian online software allows us to identify these SECIS elements in the deduced selenoproteins by introducing the file obtained from fastasubseq command.
Moreover, Seblastian can deduce selenoproteins upstream SECIS elements. Therefore, the obtained results in Seblastian were later compared with the results obtained by running our program and using the gathered results a conclusion was deduced.
CLUSTAL OMEGA
Finally, we realised that sometimes in selenoprotein families the same scaffold was aligned with different queries of the family with great identity and e-values. Therefore, in order to decide which scaffold had to be assigned to each protein we generated a phylogenetic tree and a multiple alignment with all the queries of the family and the aligned scaffolds using ClustalOmega server (https://www.ebi.ac.uk/Tools/msa/clustalo/)).
On one hand, the phylogenetic tree allowed us to asses the evolutionary relationship between the human queries and the aligned scaffolds. On the other hand, the multiple alignment showed the coverage of each scaffold for the different queries.
This way, the scaffold that was evolutionary closer to the query in the phylogenetic tree and at the same time showed a great coverage was determined to be the homolog protein in question in the genome of Craseonycteris thonglongyai.
At the same time, the phylogenetic trees were used to try to understand the evolution of proteins from the different families and to compare our results with other studies.