what we have done
MATERIALS & METHODS
Algorithm creation
In order to characterize the selenoproteome of Callopanchax toddi and its machinery, an algorithm in perl language containing all the following methodology was created in the emacs GUI text editor. The folder containing all is needed to run the programme must be named Callopanchax_toddi. It must also be at the same directory as the terminal for the algorithm to work.
Scaffold pool creation
SelenoDB 2.0 and UniProt annotated selenoproteins of Danio rerio were used as a query and compared with the genome scaffolds of Callopanchax toddi (extracted from /mnt/NFS_UPF/soft/genomes/2019/Callopanchax_toddi/). The analysis included selenoproteins and Cys-containing homologues (DIO1/2/3a/3b, GPx1a/1b/2/31/3b/4a/4b, MSRB1a/b, Sel15, SELENOE, SELENOH, SELENOI, SELENOJ1, SELENOK, SELENOL, SELENOM, SELENON, SELENOO1/O2, SELENOP, SELENOS, SELENOT1/T1B/T2, SELENOU1A, SELENOW,, TXNRD2/3), as well as selenoprotein machinery (SEPHS1/2 or SPS1/2, PSTK, SBP2, SECp43, SecS, eEFsec). If there were isoforms, the largest one was chosen.
Tblastn was used to compare the scaffolds of Callopanchax toddi with the queries. Special symbols were removed and “U” amino acids (Selenocysteine) needed to be changed to “X” to run the programme. Hits with a lower E-value than 0.01 were selected. The scaffolds where is more likely to find them were used in the following methodology.
Scaffold analysis
From the pool of selected scaffolds, they were extracted one by one using the fastafetch command and the genome index (/mnt/NFS_UPF/bioinfo/BI/genomes/2018/Callopanchax_toddi/genome.index). Extraction of target sequences -fastasubseq- and posterior multi-exonic gene model -exonerate- were used by Seblastian to find SECIS elements, while fastaseqfromGFF and fastatranslate provided protein prediction.
Alignment methods
T-coffee aligned amino acid predicted sequences with the query proteins. The best score regarding relationship between scaffold length and coverage -over 60% for good quality-, together with other criteria -similar length of the query and the predicted protein, SECIS prediction (3’-UTR region and upstream the same strand), the presence of selenoprotein residues, phylogenetic proximity-, indicated what genes encode different selenoproteins.
Phylogenetic tree
Phylogenetic trees created with phylogeny.fr were used to study the relationship between the the query and both the different scaffolds for each protein and proteins within the same family. Scaffolds closer to the query were used to find selenoprotein genes, its machinery, and SECIS elements.
General scheme
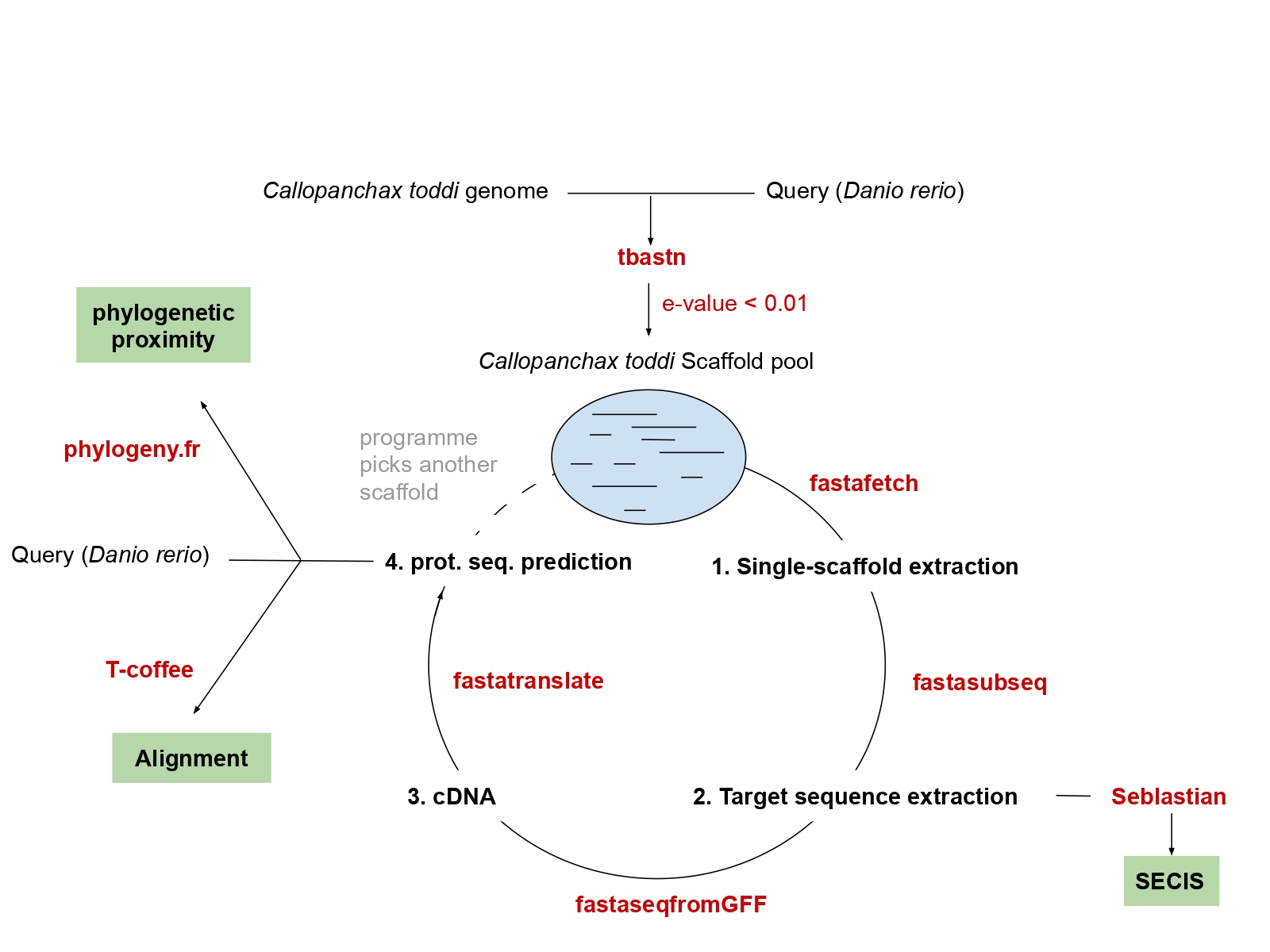
Figure 1. General procedure of the algorithm that allows finding selenoproteins within the genome of Callopanchax toddi.