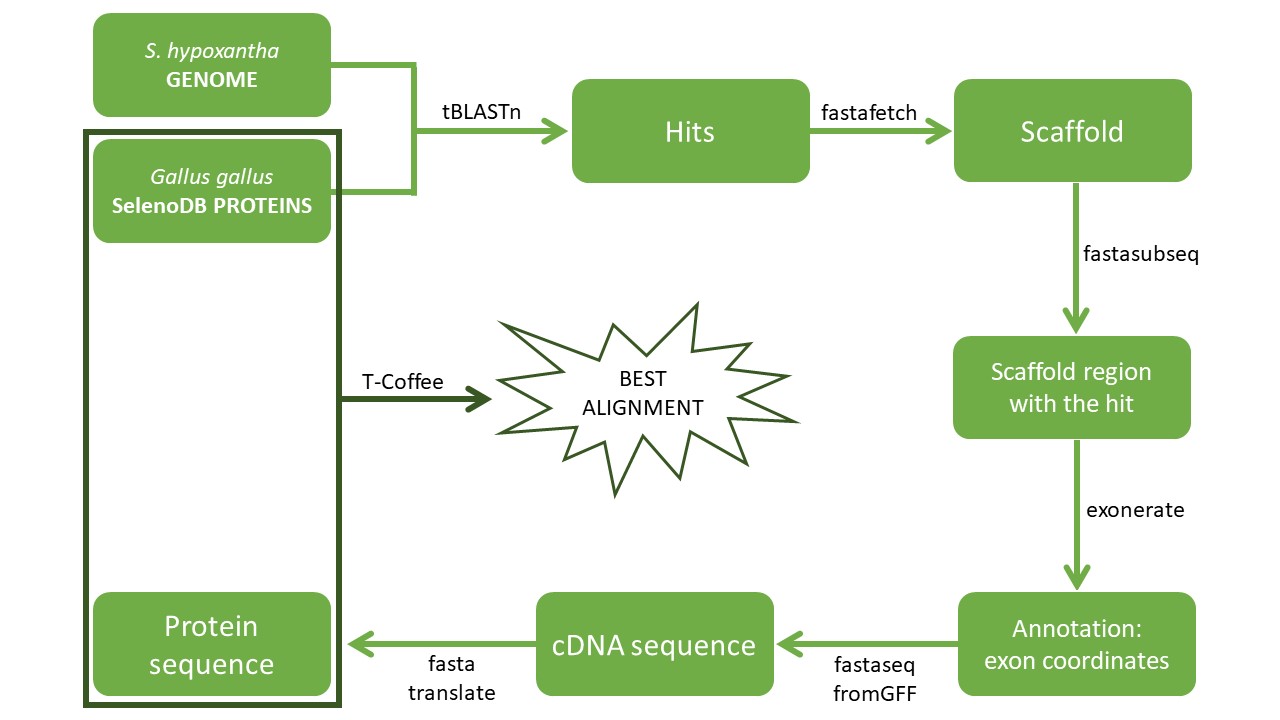
The aim of this project was to identify and annotate the selenoproteins and other related genes of Sporophila hypoxantha. Thus, the genome of the chicken (Gallus gallus) was used as the reference genome because of its phylogenetic proximity to S. hypoxantha. However, the human's (Homo sapiens) was used as a reference genome as well because the chicken selenoproteins were not as accurately-annotated as the human one. Therefore, the following procedure with the chicken was performed again with the human as reference genome. Here is an overview of the steps:
S. hypoxantha genome download
The genome of S. hypoxantha was provided through the following directory as a FASTA file:
/cursos/20428/BI/genome/2017/Sporophila_hypoxantha/genome.fa
Download of selenoproteins and other related proteins of the chicken and human
Selenoproteins and other related proteins' FASTA files from the chicken were downloaded from the selenoproteins database SelenoDB 2.0: chicken.fa. Selenoproteins are indicated in orange and Cys-containing homologs or selenium machinery genes in green here. As for the human, the SelenoDB 1.0 database was used as it is more accurate than the SelenoDB 2.0 database, thus assuring a more precise annotation of the protein: human.fa. Selenoproteins are indicated in orange, Cys-containing homologs in green, other amino acid-containing homologs in yellow and selenium molecular machinery in brown here. Before continuing, Us in the FASTA file were replaced by Xs.
Software upload
First, we need to install the following software in order to proceed:
- module load modulepath/goolf-1.7.20
- module load BLAST+/2.2.30-goolf-1.7.20
- module load T-Coffee/11.00.8cbe486-goolf-1.7.20
- module load Exonerate/2.2.0-goolf-1.7.20
- export PATH=/cursos/20428/BI/bin:$PATH
- export PATH=/cursos/20428/BI/soft/genewise/x86_64/bin:$PATH
- export WISECONFIGDIR=/cursos/20428/BI/soft/genewise/x86_64/wise2.2.0/wisecfg/
tBLASTn
With tBLASTn we localize the region where the genes of the chicken selenoproteins are found in the database of the genome of S. hypoxantha:
tblastn -query filequery.fa -db nameBLASTddbb -out outputfile
Where filequery.fa is the multi-FASTA file that contains the chicken selenoproteins and other related proteins, nameBLASTddbb is the name of the BLAST database in which we want to do the search, and outputfile is the name of the file in which we want BLAST to storage the results of the search.
In our case:
tblastn -query chicken.fa -db genome.fa -evalue 0.05 -outfmt 7 -out testblast
NOTE: We decided to limit the search by specifying that we wanted an e-value lower than 0.05, as well as we wanted the output to have a tabular format with comment lines by using -outfmt 7.
Here are the output files of the tBLASTn: testblast_chicken for the chicken and testblast_human for the human.
Once the search was done, we selected the scaffolds of interest based on the percentage of identity, the e-value and the lenght.
Semi-automation
The next steps were automated: programa.pl. However, before running it each protein was put in a file alone as the program was made for a single protein. The program includes:
Fastafetch
Fastafetch extracts the genomic sequence (scaffold) that contains the hit/s previously found with tBLASTn. In order to use Fastafetch, we need the genome structured by an index, which was previously codified and provided through the following directory:
/cursos/20428/BI/genomes/2017/Sporophila_hypoxantha/genome.index
The command used was:
$fastafetch /cursos/20428/BI/genomes/2017/Sporophila_hypoxantha/genome.fa /cursos/20428/BI/genomes/2017/Sporophila_hypoxantha/genome.index scaffold_name > protein.scaffold.fastafetch
Where:
- - scaffold_name is the elected scaffold
- - protein.scaffold.fastafetch is the output file
Fastasubseq
Fastasubseq extracts the region or subsequence of the single sequence (scaffold) already extracted with Fastafetch that contains our best hits, which were aligned in the blast.
The command used was:
$fastasubseq protein.scaffold.fastafetch start lenght > protein.scaffold.fastasubseq
Where:
- - start is the first nucleotide in the query
- - lenght is the distance between the start nucleotide and the last one in the query
- - protein.scaffold.fastasubseq is the output file
The start and end of the hit were extended 50,000 nucleotides per site, in order to obtain a region of interest that contained the whole gene, and always taking into account that the start must be higher than 1 and the length shorter than the contig's length.
Exonerate
Exonerate generates an annotation by extracting the genomic region -meaning the exons- that potentially contains the gene we are looking for in the genome of S. hypoxantha. In order to obtain directly the positions of the exons and not have to run Exonerate twice, we piped the command egrep to the Exonerate command.
The commands used was:
$exonerate -m p2g --showtargetgff -q protein_name -t protein.scaffold.fastasubseq --exhaustive yes > protein.scaffold.exon.gff
$egrep -w exon protein.scaffold.exon.gff > proteina.scaffold.egrep.gff
Where:
- - m p2g makes possible an alignment between a genome and a protein
- - showtargetgff gives all the information in a GFF file format
- - q refers to the query (Gallus gallus)
- - t refers to the target (Sporophila hypoxantha's region of interest)
- - --exhaustive yes allows us to make a more exhaustive search
- - egrep -w exon extracts all the exon regions found
- - protein.scaffold.exon.gff is the output file of Exonerate
- - protein.scaffold.egrep.gff is the output file of egrep
FastaSeqFromGFF
FastaSeqFromGFF obtains the cDNA sequence that encodes the final protein.
The command used was:
$fastaseqfromGFF.pl protein.scaffold.fastasubseq protein.scaffold.exon.gff > protein.scaffold.nt
Where:
- - protein.scaffold.nt is the output file
Fastatranslate
Fastatranslate translates the coding sequence from nucleotides to aminoacids.
The command used was:
$fastatranslate -f protein.scaffold.nt -F 1 > protein.scaffold.pep
Where:
- - -F indicates 'forward' and the nucleotide position where the traduction will begin; as the protein prediction is done from cDNA (exons), we used the option -F 1
- - protein.scaffold.pep is the output file
T-Coffee
T-Coffee makes a global alignment between a selenoprotein of the chicken and our annotation of the predicted protein of the tawny-bellied seedeater.
The command used was:
$t_coffee protein.scaffold.pep protein.fa > protein.scaffold.final.txt
Where:
- - protein.fa is the name of the protein of reference (chicken in this case)
- - protein.scaffold.final.txt is the output file
However, the Fastatranslate inserted an asterisk (*) in the Sec position in those Sec-containing sequences in the S. hypoxantha genome. Thus, we switched manually the asterisk to an X as T-Coffee cannot read asterisks. Finally, the T-Coffee was run again.
Genewise
Some T-Coffee outputs had bad alignments, so we decided to run Genewise -alternative to Exonerate- in order to generate another gene prediction to determinate if the alignment could improve using another algorithm. Nonetheless, the new T-Coffee outputs were similar or even worse than the previous ones. In this manner, we decided not to use this latter outputs to analyse the results.
Seblastian
To corroborate if the predictions made were selenoproteins, SECIS elements were predicted in the corresponding sequence. The SECIS elements are fundamental for the synthesis of selenoproteins. They are found in the 3'UTR region and direct the cell to translate UGA codons -which is normally a stop codon- as selenocysteines. In order to carry out this last step, we used the program SECISearch3/Seblastian. The option to obtain all the SECIS elements was chosen in order to select the best option. In the server, we introduced the sequence obtained in the Fastasubseq output file. However, as the genome of S. hypoxantha lacked a high degree of accuracy: in some positions, random characters were found instead of nucleotides. As a consequence, we changed these characters to N's by using the sed command, which is included in the programa.pl previously mentioned in the semi-automation section.