
Materials i Mètodes
Selecció de les proteïnes a analitzar
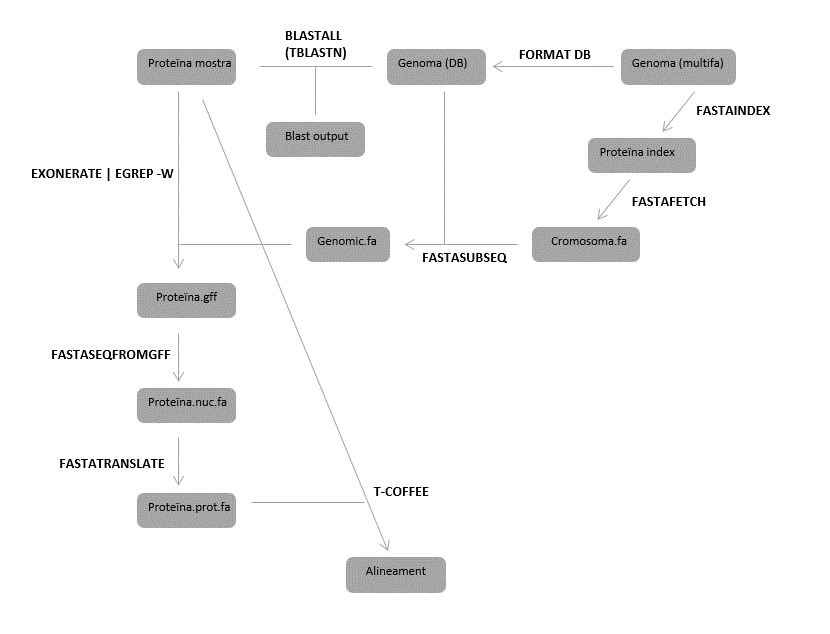
El primer pas a realitzar és mirar quines són les selenoproteïnes descrites en espècies de referència. S'ha escollit l'humà (Homo sapiens) i el pollastre (Gallus gallus). Per tal de poder identificar les selenoproteïnes al Struthio camelus australis i anotar la seva posició al genoma, s'ha seguit un procés en el que en cada apartat s'ha utilitzat un programa diferent. El primer que s'ha de fer és exportar els camins per tal de poder utilitzar alguns dels programes del procés. Les comandes que s'utilitzen són:
$ export PATH=/cursos/BI/bin:$PATH
$ export PATH=/cursos/BI/bin/ncbiblast/bin:$PATH
$ cp /cursos/BI/bin/ncbiblast/.ncbirc ~/
$ export PATH=/cursos/BI/soft/exonerate/i386/bin:$PATH
$ export PATH=/cursos/BI/soft/t_coffee/i386/bin:$PATH
Obtenció del genoma
El genoma de l'estruç va ser donat pels professors de l'assignatura de bioinformàtica. Es troba com a fitxer multifasta al directori
/cursos/BI/genomes/vertebrates/2014/Struthio_camelus_australis/genome.fa
El primer pas que cal fer és transformar aquest fitxer multifasta a un format de base de dades. Per això s'utilitza el programa format db, amb la següent comanda al shell, i es guarda el fitxer com a estruç.fa
$ formatdb -i /cursos/BI/genomes/vertebrates/2014/Struthio_camelus_australis/genome.fa -p F -n estruç.fa
Després d'això, s'ha d'indexar el genoma, de manera que quedin ordenats els diferents fragments i Scaffolds. Amb la següent comanda s'indexa el genoma i es guarda com a estruç.index
$ fastaindex /cursos/BI/genomes/vertebrates/2014/Struthio_camelus_australis/genome.fa estruç.index
Obtenció de les querys
Per tal de trobar totes les selenoproteïnes de Struthio camelus australis, s'han enfrontat selenoproteïnes conegudes d'una espècie de referència amb el genoma de l'estruç. La seqüència de les proteïnes s'ha trobat a la base de dades d'internet SelenoDB. S'ha vist que les selenoproteïnes descrites en aus són pràcticament les mateixes que en humans, pel que s'ha decidit utilitzar les selenoproteïnes d'Homo sapiens, degut a que el seu genoma està molt ben anotat. En el cas que alguna proteïna no tingués un bon alineament, també s'han buscar proteïnes de pollastre (Gallus gallus), ja que al ser una au, el seu genoma és més proper evolutivament a l'estruç que a l'humà.
Comparació de la seqüència de la proteïna amb el genoma
Per tal d'enfrontar la proteïna amb el genoma, s'ha utilitzat el programa BLAST (Basic Local Alignment Search Tool). BLAST és un algoritme heurístic que troba regions de similaritat entre seqüències. Compara seqüències de nucleòtids o proteïnes amb una base de dades i calcula la significança estadística dels resultats trobats. El fet d'utilitzar un algoritme heurístic pot fer que es perdin alguns resultats o hits que no tinguin un grau de similitud molt alt.
En aquest treball s'ha utilitzat la versió tblastn de BLAST, que compara seqüències de proteïnes amb una base de dades de nucleòtids. Com a query s'utilitza la seqüència d'aminoàcids d'una selenoproteïna d'una espècie de referència i com a base de dades el genoma de l'estruç (estruç.fa). La comanda utilitzada és
blastall -p tblastn -i query.fa -d estruç.fa -o fitxerdesortida.blast
on -p designa quin tipus de programa de BLAST s'utilitza (tblastn), -i designa la query, -d designa la base de dades per a comparar i -o el fitxer de sortida amb els resultats.
Per analitzar els diferents hits del BLAST, cal fixar-se en el E-value i l'SCORE. Com més alt sigui l'score i més petit l'E-value, menys probabilitats hi ha que l'alineament es doni per atzar. També cal fixar-se en si la selenocisteïna de la proteïna està dins de l'alineament.
Selecció de l'Scaffold
Una vegada s'ha decidit quins hits del BLAST tenen un bon alineament, s'ha d'extreure l'Scaffold del fitxer multifasta inicial, per poder seguir amb l'anàlisi. S'utilitza la comanda
$ fastafetch /cursos/BI/genomes/vertebrates/2014/Struthio_camelus_australis/genome.fa estruç.index "Scaffold" > Scaffold.fa
on es mostra el camí fins al genoma, el fitxer on està el genoma indexat i l'identificador de l'Scaffold seleccionat.
Obtenció de la subseqüència
A partir dels hits del BLAST s'extreu un fragment de l'Scaffold seleccionat per tal d'obtenir una regió més acotada que inclogui el gen de la selenoproteïna. Per tal de fer-ho, cal ampliar la seqüència de nucleòtids del hit, per estar segurs de que no es perd cap exó. La comanda per a obtenir la subseqüència és
$ fastasubseq Scaffold.fa start length > genomic.fa
on s'indica el fitxer que conté l'Scaffold, el nucleòtid inicial de la subseqüència que es vol obtenir i l'allargada de la subseqüència en número de nucleòtids. El fragment es guarda en un fitxer amb el nom genomic.fa.
Predicció d'exons
Exonerate
Una vegada s'ha obtingut la regió del genoma que conté el possible gen, s'utilitza el programa exonerate per tal d'alinear la query amb la regió i fer una predicció de l'estructura d'un gen en aquella regió. Aquest programa detecta possibles llocs d'splicing a més d'exons i introns dins la seqüència. Exonerate no reconeix la lletra U com un aminoàcid, per tant s'ha de canviar a la seqüència de la query per una X, ja que sinó donaria error. La comanda utilitzada és
$ exonerate -m p2g --showtargetgff -q query.fa -t genomic.fa --exhaustive yes | egrep -w exon > exonerate.gff
on -m p2g indica el model de l'alineament proteïna contra genoma, --showtargetgff fa que el resultat es guardi en format gff. S'introdueix també a -q el fitxer de la query amb la seqüència de la proteïna de l'espècie de referència i a -t el fitxer genomic.fa amb la regió genòmica. --exhaustive yes permet que la predicció sigui més acurada per tal d'obtenir un resultat millor. Per tal d'extreure només els exons utilitzem la comanda egrep -w exon. Aquesta escull les línies del fitxer gff on hi ha la paraula exó i les extreu en un fitxer de format gff, que contindrà les regions exòniques predites.
Fastaseqfromgff
El programa fastaseqfromgff parteix del fitxer de sortida de l'exonerate, que està en format gff i obté la seqüencia dels exons en un fitxer en format fasta. S'utilitza la següent comanda
$ fastaseqfromGFF.pl genomic.fa exonerate.gff > protein.nuc.fa
on s'indica la regió genòmica obtinguda pel fastasubseq i les regions exòniques predites per l'exonerate. El resultat és un fitxer fasta que conté la seqüència de nucleòtids del gen predit.
Fastatranslate
Amb aquest programa es pot traduir el fitxer que conté la seqüència de nucleòtids del gen predit per tal d'obtenir un altre fitxer que contingui la seqüència d'aminoàcids de la proteïna. La comanda que cal utilitzar és
$ fastatranslate -f protein.nuc.fa -F 1 > protein.prot.fa
on a -f s'indica el fitxer de nucleòtids que es vol traduir i -F indica quin patró de lectura es vol utilitzar (al posar 1 comença a traduir a partir del primer nucleòtid de la seqüència). El resultat s'emmagatzema en un fitxer com a seqüència d'aminoàcids.
En el fitxer de la proteïna predita, el programa fastatranslate no introdueix U en trobar una selenocisteïna, sinó que posa "*" en trobar un codó STOP al mig de la seqüència. Per tant, s'han de canviar els "*" per una X per tal que funcioni la resta del procés.
T-Coffee
Finalment, el programa T-Coffee alinea la proteïna predita a partir de la seqüència genòmica de l'estruç amb la query inicial de l'espècie de referència. La comanda és
$ t_coffee query.fa protein.prot.fa > alineament.fa
on s'indica tant el fitxer de la query com el de la proteïna predita. El resultat es guarda en un fitxer en format fasta que conté l'alineament.
Automatització
Per tal de facilitar i agilitzar la feina, s'ha realitzat un programa que executa de manera automàtica el passos de l'anàlisi de proteïnes des de l'exonerate fins al T-Coffee. També s'ha escrit un programa en perl que canvia els asteriscs d'un text per X, ja que el programa T-Coffee no entén el símbol "*" i dóna error.
Per tal que els programes funcionin, cal haver exportat els camins a tots els programes i alhora permetre executar-los mitjançant la comanda de UNIX
chmod u+x programa.pl
on programa.pl és el programa que es vol executar.
Detecció d'elements SECIS
Els elements SECIS són unes estructures tridimensionals, que es troben a l'extrem 3' del gen de la selenoproteïna i són necessaris per a la seva traducció. La cerca d'elements SECIS s'ha fet a través del programa SECISearch3 utilitzant com a seqüència el fitxer amb la subseqüència obtinguda amb el programa fastasubseq.
Seblastian
Seblastian és un programa que prediu una selenoproteïna a partir d'una seqüència i la compara amb altres espècies per veure si hi ha homologia. S'ha utilitzat el mateix fitxer de la subseqüència que per la detecció d'elements SECIS.
Predicció de tRNA
S'ha buscat si en el genoma de l'estruç també hi ha la seqüència que codifica per al tRNA de la selenocisteïna. Per fer-ho, primer de tot s'ha buscat la seqüència del tRNA en les espècies de referència d'Homo sapiens i pollastre (Gallus gallus) a la base de dades Genomic tRNA Database.
Una vegada ja s'han obtingut les seqüències, s'ha realitzat un BLAST de la proteïna contra el genoma de l'estruç, per tal de detectar els Scaffolds on hi hagi un possible tRNA. La comanda que cal posar és
blastall -p blastn -i querytRNA.fa -d estruç.fa -o fitxerdesortida.blast
on la query és la seqüència del tRNA de referència i la base de dades utilitzada és el genoma de l'estruç.
Per tal de seleccionar l'Scaffold, es fa un fastafetch a partir del genoma en format multifasta. La comanda necessària és
$ fastafetch /cursos/BI/genomes/vertebrates/2014/Struthio_camelus_australis/genome.fa estruç.index "Scaffold" > scaffold.fa
on mostrem el camí fins al genoma de l'estruç, el fitxer del genoma indexat i l'Scaffold que es vol extreure.
Una vegada s'han obtingut els Scaffolds, s'utilitza el programa ARAGORN que detecta totes les seqüències corresponents a tRNA en una seqüència donada de 15Mb com a màxim.