Selenoproteïnes en Eptesicus fuscus
Cebrián M.C, Franco M, Gallardo P, Gallego E.
| ABSTRACT | INTRODUCCIÓ | MATERIALS I MÈTODES | RESULTATS | DISCUSSIÓ | CONCLUSIONS | REFERÈNCIES | AGRAÏMENTS | CONTACTA'NS |
OBTENCIÓ DEL GENOMA DE Eptesicus fuscus
Aquest genoma l'hem obtingut gràcies als professors de la assignatura mitjançant l'accés a la base:
/cursos/BI/genomes/project_2014/Eptesicus_fuscus/genome.fa
| /cursos/BI/genomes/project_2014/Eptesicus_fuscus/genome.fa |
OBTENCIÓ DE GENOMES MODEL
Les seqüències aminoacídiques emprades han sigut de la espècies més properes trobades dins de la base de dades de SelenoDb, Myotis lucifugus. Hi ha molts casos en els quals les selenoproteïnes no estaven ben anotades, ja que no començaven per metionina i per tant, hem utilitzat les de l'Homo sapiens ja que és l'espècie on les selenoproteïnes estan millor predites. Hi ha hagut seqüències on hem dubtat entre quina d'aquestes escollir, i llavors hem fet servir T-Coffee per alinear les seqüències tant de Myotis lucifugus i Homo sapiens i així escollir la més adient pel posterior anàlisi.
CERCA DE LES SELENOPROTEÏNES
Abans de començar a treballar s'han d'exportar els diferents programes perquè es puguin utilitzar.
COMANDA EMPRADA PROGRAMES
$ export PATH=/cursos/BI/bin/ncbiblast/bin:$PATH NCBI Blast $ cp /cursos/BI/bin/ncbiblast/.ncbirc ~/ NCBI Blast $ export PATH=/cursos/BI/soft/exonerate/i386/bin:$PATH Exonerate $ export PATH=/cursos/BI/bin:$PATH Genewise, fastaseqfromGFF.pl i T-coffee
$ export WISECONFIGDIR=/cursos/BI/soft/wise-2.2.0/wisecfg Genewise
| COMANDA EMPRADA | PROGRAMES |
| $ export PATH=/cursos/BI/bin/ncbiblast/bin:$PATH | NCBI Blast |
| $ cp /cursos/BI/bin/ncbiblast/.ncbirc ~/ | NCBI Blast |
| $ export PATH=/cursos/BI/soft/exonerate/i386/bin:$PATH | Exonerate |
| $ export PATH=/cursos/BI/bin:$PATH | Genewise, fastaseqfromGFF.pl i T-coffee |
| $ export WISECONFIGDIR=/cursos/BI/soft/wise-2.2.0/wisecfg | Genewise |
BLAST: UTILITZEM tBlastn
BLAST (Basic Local Alignment Search Tool) és un programa informàtic que ens alinea diferents seqüències de forma local, ja siguin de nucleòtids o aminoàcids. Existeixen diferents tipus de BLAST i nosaltres hem utilitzat tBLASTn, ja que volem comparar la nostra seqüència de proteïnes, en aquest cas la query triada després del corresponent T-Coffee, amb una base de dades del genoma d' Eptesicus fuscus.
BLAST mostra els alineaments de les seqüències i a més, calcula la significança estadística dels resultats obtenint un valor d'E-value diferent per a cada alineament fet. Tots els hits amb un E-value major a 10-5 s'han descartat i s'ha triat el que tingués el millor E-value comparant els diferents E-values de cada scaffold en cada proteïna de la mateixa família, ja que contra més petit és aquest valor, més significatiu és l'alineament trobat. A més, també s'ha utilitzat com a criteri de selecció el score del BLAST i el tant per cent d'identitat.
Per executar el tBlastn necessitem la següent comanda:
| $ blastall -p tblastn -i proteina_query .fa -d /cursos/BI/genomes/project_2014/Eptesicus_fuscus/genome.fa -o fitxersortida |
L'argument -p s'indica quin tipus de BLAST estem emprant. La -i indica la ubicació de la proteïna (query) i està en format FASTA, mentre que la -d indica la ubicació de la base de dades del genoma (Eptesicus fuscus) i -o serà el fitxer de sortida on s'emmagatzemaran els resultats del BLAST. Al programa hem limitat els hits que es poden obtenir utilitzant l'argument -e i el -m perquè apareguin els resultats en una taula i així fos més fàcil l'anàlisi posterior.
EXTRACCIÓ DE LA REGIÓ GENÒMICA DELS HITS SELECCIONATS MITJANÇANT EXONERATE
- Fastaindex
-
El que fa la següent comanda és organitzar el fitxer del genoma en diferents regions per després poder agafar la regió que interessa amb el Fastafetch.
- Fastafetch
- Ara realitzem un fastafetch per extreure l'scaffold on es troba cada un dels hits significatius trobats anteriorment amb la següent comanda:
- Fastasubsequence
- Al final seleccionem la regió que envolta el gen que estem buscant i així obtenim una seqüència més curta amb la que treballarem a continuació. Hem agafat les posicions dels hits obtinguts al tBLASTn i expandim els marges 50.000 nucleòtids del gen upstream i downstream. Tot i això, en els casos en què el resultat de l’Exonerate Exhaustive posterior començava massa lluny de l’inici de la proteïna (i per tant, de la Metionina) allargàvem un major nombre de nucleòtids per obtenir uns millors resultats. Utlitzem la següent comanda:
- Exonerate
- Aquest programa ens permet obtenir un alineament molt més acurat que el BLAST i, a més, ens prediu l'estructura exònica de la seqüència problema. És a dir, alinea la seqüència de la selenoproteïna de l'organisme model amb una regió genòmica extreta del nostre organisme gràcies a fastasubseq. Això ens diu si el hit obtingut dins l'exó codifica per una proteïna o no. Utilitzem la següent comanda:
| $ fastaindex /cursos/BI/genomes/project_2014/Eptesicus_fuscus/genome.fa genome.index |
S'indica la ubicació del genoma i just després la ubicació del fitxer de sortida on estarà tot indexat.
$ fastafetch /cursos/BI/genomes/project_2014/Eptesicus_fuscus/genome.fa genome.index "scaffold" > fastafetch.fa
| $ fastafetch /cursos/BI/genomes/project_2014/Eptesicus_fuscus/genome.fa genome.index "scaffold" > fastafetch.fa |
Genome.index és el fitxer on tenim indexat el genoma i "scaffold" són els scaffolds obtinguts al tBlastn i fastafetch.fa serà el nom de la ubicació del fitxer de sortida.
$ /cursos/BI/soft/exonerate/i386/bin/fastasubseq fastafetch.fa> start length > genomic.fa
Start és el nuclèotid amb el que comencem la subseqüència, length és la llargada dels nucleòtids que volem extreure i genomic.fa és el nom del la ubicació del fitxer de sortida.
| $ /cursos/BI/soft/exonerate/i386/bin/fastasubseq fastafetch.fa> start length > genomic.fa |
| $ exonerate -m p2g --showtargetgff -q proteina_query.fa -t genomic.fa > exonerate.gff |
-m indica el tipus d'alineament (p2g indica proteïna contra genoma), –showtargetgff indica que el resultat es mostri en format gff i -q indica la ubicació de la proteïna. Finalment, la -t indica la regió del genoma amb el gen d'interès. A continuació ho redireccionem en un arxiu en format GFF.
Hem afegit a la comanda l'opció -E (exhaustive) per fer cerques més acurades en els casos on calia, ja que a vegades l'exonerate et donava un alineament que no començava per la posició 1.
| $ exonerate -m p2g --showtargetgff -q proteina_query.fa -t genomic.fa -E > exonerate.gff |
Ara utilitzem la següent comanda per a seleccionar només els exons.
| $ egrep -w exon exonerate.gff > exonerateexon.gff |
Egrep selecciona les línies on apareix el patró que definim dins l'argument -w (exon en aquest cas). Exonerate.gff és el fitxer GFF que utilitzem i exonerateexon.gff és el fitxer de sortida on només tindrem els exons.
Ara utilitzem fastaseqfromGFF.pl per obtenir el cDNA en format FASTA amb la següent comanda:
| $ fastaseqfromGFF.pl genomic.fa exonerateexon.gff > cDNA.fa |
Genomic.fa és la subseqüència extreta anteriorment, cDNA.gff és l’arxiu en format gff que només conté els exons i cDNA.fa és el fitxer de sortida que contindrà la seqüència del cDNA en format FASTA.
Ara executem fastatranslate que tradueix el cDNA a proteïna, obtenint sis possibles marcs de lectura (ORFs) que són les 3 directes i 3 reverse i ho guardem en un arxiu que es diu translate.fa.
| $ fastatranslate cDNA.fa > translate.fa |
Mirem l'arxiu i en la majoria dels casos decidim afegir -F 1 per a què ens agafi només la primera pauta de lectura forward, ja que és la pauta real (comença per metionina o bé per la seqüència obtinguda per l'Exonerate). Per tant, emprem la següent comanda:
| $ fastatranslate -F 1 cDNA.fa > aa_gen.fa |
aa_gen.fa serà l'arxiu on estarà la primera pauta de lectura.
GENEWISE
Genewise és un altre programa de predicció de gens, amb un mateix objectiu tot i utilitzar algorismes diferents. L'hem fet servir per verificar els resultats obtinguts amb el programa Exonerate i per comparar si els dos programes donaven resultats semblants. Se sap que aquest programa fa un alineament més precís que el BLAST entre la proteïna problema (query) i la regió genòmica observada.
| $ genewise -pep -pretty -cdna -gff proteina_query.fa genomic.fa > genewise.gff |
-Pep obtindrem la seqüència peptídica predita en el fitxer de sortida, -pretty és per a què ens mostri l'alineament, -cdna és per a què ens mostri la seqüència genòmica alineada, -gff per a què ens mostri la informació en format GFF, proteina_query.fa és la ubicació de la nostra proteïna problema, genomic.fa és la subseqüència extreta anteriorment i genewise.gff és el fitxer de sortida. Si hem d'executar el Genewise de la cadena reversa hem d'afegir l'argument -trev:
| $ genewise -pep -trev -pretty -cdna -gff proteina_query.fa genomic.fa > genewise.fa |
T-COFFEE
Després d'haver obtingut les seqüències de les proteïnes amb els mètodes Exonerate i Genewise emprem el programa T-Coffee per tal d'alinear la proteïna obtinguda amb la query inicial. Podrem veure si els residus d'aminoàcids de la proteïna problema i la proteïna predita es corresponen. Executem la següent comanda si agafem la proteïna predita pel programa Exonerate:
| $ t_coffee proteina_query.fa aa_gen.fa > tcoffee.fa |
Aquesta comanda també la realitzarem amb la proteïna predita pel Genewise, però primer hem d'agafar-la de l'arxiu genewise.fa i guardar-la en un arxiu que anomanarem aa_seqgenewise.fa i executem aquesta comanda:
| $ t_coffee proteina_query.fa aa_seqgenewise.fa > tcoffeegenewise.fa |
SECISEARCH
Ara un cop hem finalitzat la cerca de selenoproteïnes per saber si aquestes tenen elements SECIS, executem el programa SECIsearch.pl, encara que també es pot fer mitjançant la seva pàgina web.
Aquest programa ens permetrà confirmar si el codó UGA trobat a la selenoproteïna correspon a una selenocisteïna i no a un codó stop, ja que els elements SECIS són essencials per a la incorporació d'aquest aminoàcid. Els elements SECIS reals s'han de trobar a poques bases de distància de la selenoproteïna a l'extrem 3' de la cadena. Per executar-ho emprem la següent comanda:
| $ SECISearch.pl genomic.fa > seci.fa |
genomic.fa és l'arxiu amb la subseqüència extreta emprant el programa fastasubseq.
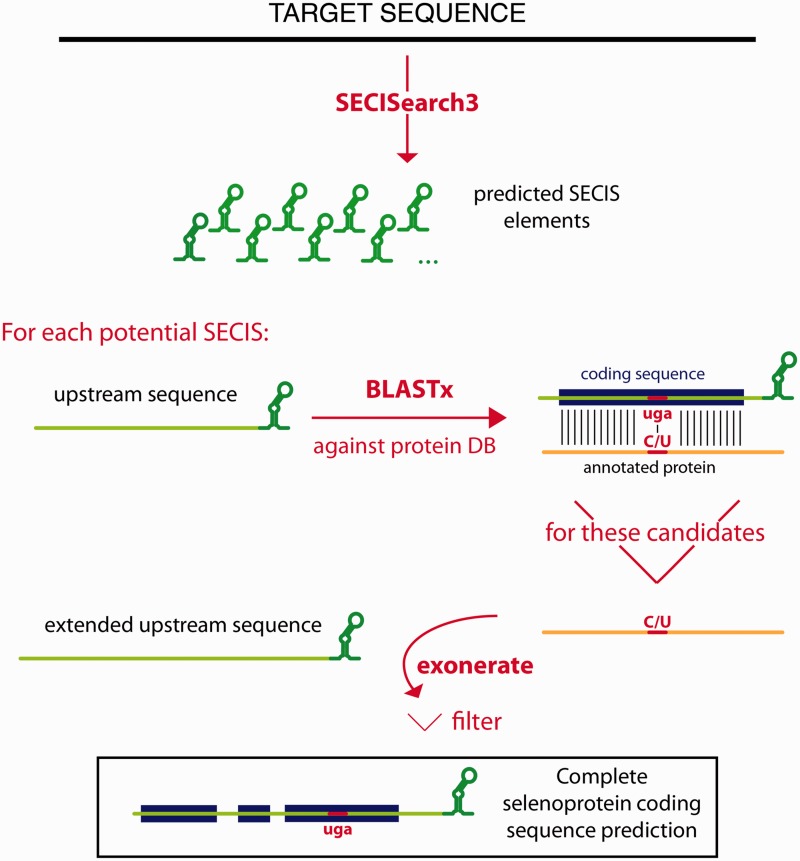
SECISEARCH3 I SEBLASTIAN
Seblastian és un programa basat en SECISearch3, que és una combinació de prediccions de Infernal, Covels i l’original SECISearch, que ens prediu la selenoproteïna.
Tots els possibles elements SECIS es prediuen en una seqüència target i la seqüència upstream de cada candidat SECIS s'examina per a veure si codifica per una possible selenoproteïna. La seqüència upstream de cada SECIS es fa córrer amb Blastx contra una base de dades de proteïnes. Les proteïnes anotades en la base de dades s'utilitzen com a proteïna problema mentre que la seqüència nucleotídica com a target. Es fan dos tipus d'alineaments de Blast:
-
Un en el qual el selenocisteïna de la proteïna problema s'alinea amb UGA de la seqüència target
Un en el qual la cisteïna de la proteïna problema s'alinea amb UGA de la seqüència target.
Aquest procediment fa que s'obtinguin dos tipus de candidats: selenoproteïnes conegudes i noves, homòlogues de selenoproteïnes ja conegudes. A continuació, fa córrer el programa Exonerate. Com a proteïna problema, s'utilitza la seqüència obtinguda en l’alineament del Blast mentre que com a target s'usa la regió en el mateix alineament del BLAST, que ha sigut apropiadament estès. Al final, tots els candidats passen un filtre. Aquests requereixen tenir l’element SECIS correctament posicionat i que no contingui pseudogens-like.
FILOGÈNIA
Per tal de poder conèixer l'scaffold adient de les diferents proteïnes presents d’una mateixa família, s’ha fet la filogènica d’aquesta família usant la pàgina web, a partir de l’apartat “Phylogeny analysis” > “One click”.
Hem introduït les diferents seqüències disponibles a SelenoDB i les seqüències de les proteïnes predites a partir de l’Exonerate. D’aquesta manera, el web ens diu quin hit és l’adient i per tant, quin hauríem d’haver usat per fer l’anàlisi. Tot i això, a l’haver realitzar la filogènia com a punt final del treball, no sempre hem tingut temps suficient per corregir els errors al moment d’escollir els hits.
Blastp
En les proteïnes en què els resultats no eren informatius (PSTK i GPX8) vam emprar el programa Blastp , el qual ens busca proteïnes dels diferents genomes de la base de dades NCBI utilitzant la proteïna predita com a query. Aleshores, obtenim proteïnes que presenten alta homologia amb la proteïna predita en altres espècies properes.
CONSIDERACIONS
Tot el procés de recerca ha estat fet de forma manual. Després de cada pas hem anat verificant els diferents fitxers per a poder fer un correcte anàlisi posterior. Durant tot el procés si hi havia algun problema l'hem corregit en el cas que fos necessari.
AUTOMATITZACIÓ
Hem creat un programa amb Perl que ens canviï totes les U per X en les selenoproteïnes per a poder dur a terme els diferents programes sense problemes posteriorment.
#!/usr/bin/perl
use strict;
my @v;
my $string;
my $seq;
my $i;
my $longitutvector;
# Reemplazamos todas las "U" por "X" print "Entra una sequencia proteica: \n";
#die("Stop here"); $seq= < STDIN >; chomp ($seq); @v = split (//,$seq);
$i = 0;
$longitutvector = scalar(@v);
while ($i < $longitutvector) {
}
$seq = join('',@v);
print "$seq\n";
I un altre programa per poder fer els Blast automàticament.
my $genome_path = '/cursos/BI/genomes/project_2014/Eptesicus_fuscus/genome.fa ';
my $proteins_path = '/home/U74303/luke/Test_selenoproteins/SEQS/';
my $blast_path = '/home/U74303/luke/Test_selenoproteins/BLAST_OUT/';
# we put the protein files into @protein_files array (lista)
# Contacte: monica.franco01@estudiant.upf.edu
opendir ( my $dir_proteins, "$proteins_path" ) || die ("Cannot open dir ${proteins_path}:$!");
my @protein_files = readdir $dir_proteins;
closedir $dir_proteins;
my $prot_name;
my $database = "$genome_path";
my $blast_out;
my $blast_command;
foreach my $file (@protein_files) {
$prot_name = $file =~ m/([a-zA-Z0-9\.\s_]+)\.fa/;
$blast_out = "$blast_path" . $1 . "_blast.out";
print "$blast_out\n";
my $blast_command = "blastall -p tblastn -i $prot_name -d $database -o $blast_out -e 0.0001 -m 8";
system($blast_command);
}