
Materials i mètodes
L'objectiu del nostre treball consisteix en identificar totes les selenoproteïnes de la tortuga Chrysemys picta bellii i anotar en el genoma on es localitzen. El procediment seguit es divideix en diferents apartats, que a la vegada es corresponen amb els diferents programes utilitzats.Programes i automatització
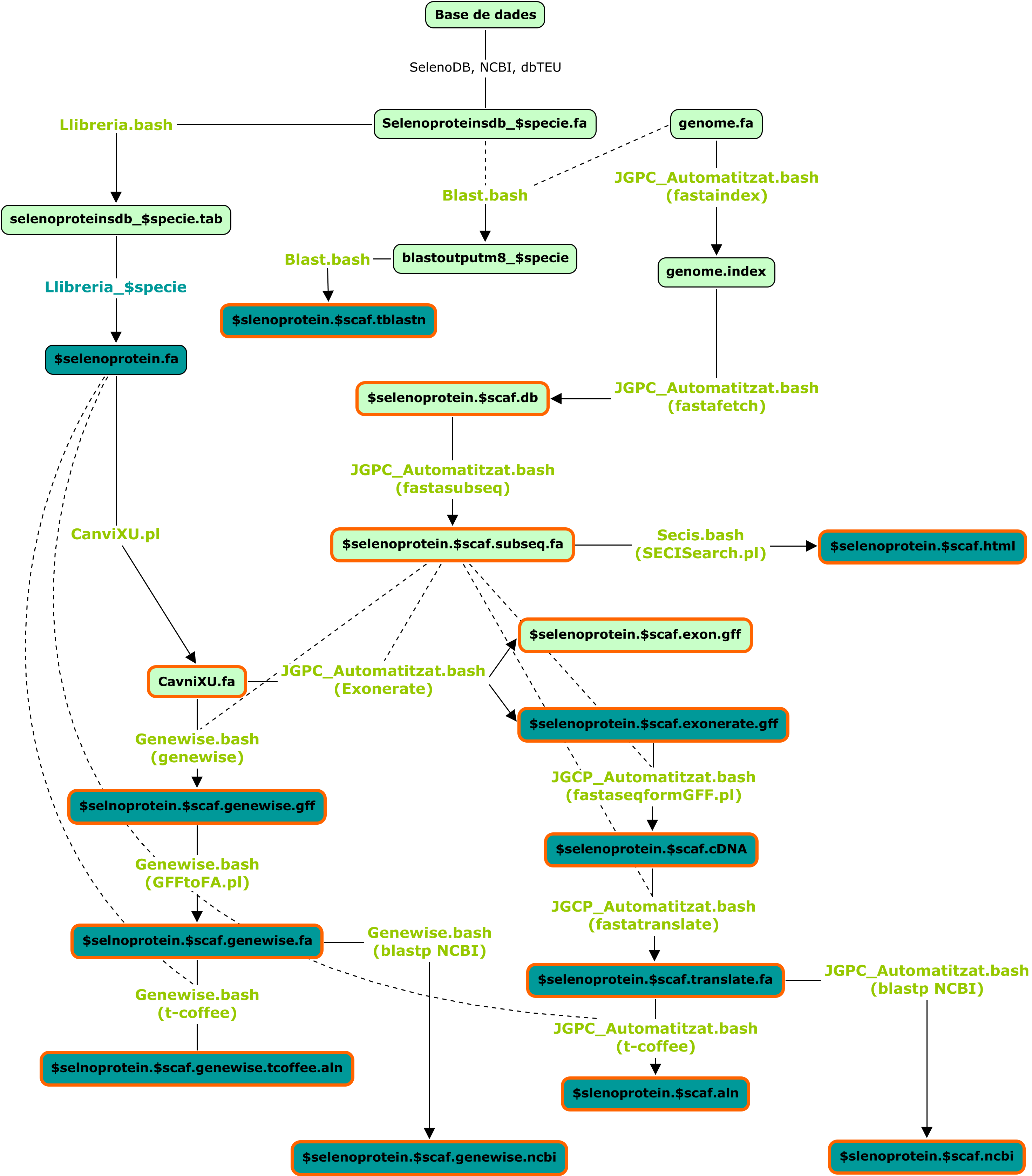
Per tal d'agilitzar tot el procés d'anàlisi i obtenció de dades, hem creat diversos programes en llenguatge bash i perl. Aquests permeten automatitzar tots els passos per anotar una selenoproteïna en el genoma de Chrysemys picta bellii i facilitar l'anàsi i la millora de les dades resultants.
Per més informació i descarregar els programes clica aquí.
Genoma i querys
Obtenció del genoma
El genoma de Chrysemys picta bellii utilitzat es troba al directori:/cursos/BI/genomes/project_2013/Chrysemys_picta_bellii
Aquest ja es troba en forma de base de dades de BLAST.
Obtenció de les querys
Per a obtenir la seqüència de les selenoproteïnes conegudes vam recòrrer a diferents bases de dades. L'anàlisi es va realitzar amb les selenoproteïnes, i la respectiva maquinària, de diferents organismes: l'humà, el pollastre i altres selenoproeïnes de Zebra fish, Fugu, Tetraodon o Xenopus. La procedència d'aquestes és:
Humà: SelenoDBPollastre: dbTEU
Zebra fish, Fugu, Tetraodon i Xenopus: NCBI
Inicialment es va escollir com a query les selenoproteïnes de l'humà, ja que en la base SeloDB és el mamífer amb més selenoproteïnes anotades. Posteriorment, vam seleccionar un nou rang de seqüències de selenoproteïnes. Aquest cop la query procedia de Gallus gallus, ja que la vam considerar l'espécie més propera filogenéticament a la nostra tortuga que tingués selenoproteïnes correctament anotades. Per últim, ens vam interessar en un nou tipus de query. Vam escollir selenoproteïnes que pertanyessin exclusivament als bonnyfishes o altres espècies que no es trobessin en humà o en pollastre, per averiguar si aquestes s'havien perdut.
Si voleu descarregar les querys utilitzades al durant el treball, aneu a Resultats
Tornar a dalt
Cerca de similaritat: tBlastn
El BLAST (Basic Local Alignment Search Tool) és un programa informàtic d'alineament de seqüències de tipus local, ja sigui de DNA o de proteïnes. El programa és capaç de comparar una seqüència problema (anomenada query) contra una gran quantitat de seqüències que es troben en una base de dades. L'algorisme que fa servir troba les seqüències de la base de dades que tenen major semblança a la seqüència query molt ràpidament, però és heurístic i per tant es poden perdre hits reals que no presenten una similaritat molt elevada. Segons la naturalesa de la nostra query (DNA o porteïna) i segons la naturalesa de la base de dades objectiu farem servir un tipus de BLAST o un altre. Nosaltres hem utilitzat tBLASTn, el qual permet comparar una seqüència proteica (query) amb la base de dades de nucelòtids d'interès.Per extreure el software necessari per la utilització d'aquest programa és necessari donar aquestes dues ordres al shell:
$ export PATH=/cursos/BI/bin/ncbiblast/bin:$PATH
$ cp /cursos/BI/bin/ncbiblast/.ncbirc ~/
Per executar una cerca tBLASTn d'una query contra una base de dades s'ha d'utilitzar la comanda següent:
$ blastall -p tblastn -i query.fa -d genoma.fa -o proteinavsgenoma.tblastn
on -p és el tipus de blast, -i és la ubicació de la query, -d és la ubicació de la base de dades i -o és el nom on s'exportarà la informació d'output.
Per tal d'automatitzar el procés, hem creat el programa blast.bash. Aquest inclou la comanda:
blastall -p tblastn -i ./selenoproteinsdb_$specie.fa -d /cursos/BI/genomes/project_2013/Chrysemys_picta_bellii/genome.fa -o ./blastoutput_$specie -e 1e-5 -m8
En aquest cas, el blast en mostrava els hits signaficatius amb un e-value de 1e-5 o inferior. Alhora, per tal d'optimitzar el procés d'automatització, vam realitzar un blast en format m8 (format columnar) a més del tblastn habitual. $specie fa refència a la procedència de les querys (humà, pollastre, peix...).
Tornar a dalt
Extracció de la regió genòmica: Fastaindex, Fastafetch i Fastasubseq
Per tal de poder extreure primer la seqüència que conté la regió del gen, i després extreure la regió genòmica de la selenoproteina a partir de la seqüència, necessitem l'ajuda d'aquest tres programes, tots tres formant part del software d'Exonerate:
Fastaindex
Organitza el genoma en diferents regions. En el cas del genoma de Chrysemys picta bellii genera un índex, anomenat genome.index, que ordena el genoma per scaffolds.
fastaindex /cursos/BI/genomes/project_2013/Chrysemys_picta_bellii/genome.fa ./genome.index
Fastafetch
Selecciona una regió indicada i la desa en un document en format db. En el nostre cas, selecciona l'scaffold.
fastafetch /cursos/BI/genomes/project_2013/Chrysemys_picta_bellii/genome.fa genome.index $scaffold > ./$selenoprotein/fastafetch/$selenoprotein.$scaf.db
$selenoprotein fa referència a la selenoproteina que estem analitzant (Ex: SPPSelM_human). $scaffold engloba el nom del scaffold seleccionat (Ex: gi|371559437|gb|JH584396.1|) i $scaf correspon al nom abreviat d'aquest últim (Ex: JH584396.1).
Fastasubseq
Selecciona una zona encara més delimitada de la regió, concretament la que envolta el gen que estem buscant. D'aquesta manera, obtenim una seqüència més curta, amb la que serà més fàcil treballar. S'ha d'agafar les posicions del hit obtingut al BLAST i expandir els marges de manera que ens assegurem la presència del gen upstream i downstream.
fastasubseq ./$selenoprotein/fastafetch/$selenoprotein.$scaf.db $start $length > ./$selenoprotein/fastasubseq/$selenoprotein.$scaf.subseq.fa
Per assegurar que en aquesta seqüència escollida hi trobem el gen d'interés, vam ampliar aquesta regió. La vam allargar 20kb a cada costat del hit obtingut al blast, que a la vedaga depenia de la longitut de la query de la que partimiem. En casos en que creiem necessària seleccionar una regió més amplia, analitzavem la selenoproteina en concret mitjançant el programa SelSearch.bash.
Tornar a dalt
Predicció d'exons: Exonerate i Genewise
Tant l’Exonerate com el Genwise són softwares que ens proporcionen un alineament més precís i una anotació del gen corresponent a la proteïna query, és a dir, obtenim una descripció detallada del gen: zones d’splicing, exons, introns, etc.
Exonerate
Exonerate ens alinea el fragment de DNA que hem extret amb el Fastasubseq amb la seqüència de la proteïna inicial. D'aquesta manera ens anota els diferents hits significatius en el genoma de la tortuga. En aquest punt vam haver de modificar el fitxer de la nostra query, canviant la U per una X o eliminant qualsevol altre símbol que Exonerate no reconeguès (Per exemple #, % o @), d'altre banda ens mostrava error. Per això, vam crear un petit programa perl anomenat CanviXU.pl que s'executava dins del programa automatizat JGPC_Automatitzat.bash Prèvia a l'execució de l'Exonerate, cal introduir la següent path:
$ export PATH=/cursos/BI/soft/exonerate/i386/bin:$PATH
L'ordre per executar el blast, inclós en el programa JGPC_Automatitzat.bash, és:
exonerate -m p2g --showtargetgff -q ./$selenoprotein/canviXU.fa -t ./$selenoprotein/fastasubseq/$selenoprotein.$scaf.subseq.fa > ./$selenoprotein/exonerate/$selenoprotein.$scaf.exonerate.gff
Fastaseqfromgff
Un cop executada la comanda obtenim un fitxer en format .gff, preparat per al processament i obtenció de la seqüència petídica. El programa fastaseqfromGFF.pl, inclós en el directori de l'assignatura (/cursos/BI/bin), ens permet extreure la seqüència del cDNA del fitxer.gff que ha generat Exonerate. Les comandes necessàries són:
exon=`grep -w exon ./$selenoprotein/exonerate/$selenoprotein.$scaf.exonerate.gff`
echo "$exon" > ./$selenoprotein/exonerate/$selenoprotein.$scaf.exonerate.exon.gff
fastaseqfromGFF.pl ./$selenoprotein/fastasubseq/$selenoprotein.$scaf.subseq.fa ./$selenoprotein/exonerate/$selenoprotein.$scaf.exonerate.exon.gff > ./$selenoprotein/cDNA/$selenoprotein.$scaf.cDNA
Fastatranslate
A continuació hem traduït el cDNA a proteïna amb el programa Fastatranslate, que acompanya a l'exonerate.fastatranslate -F 1 "./$selenoprotein/cDNA/$selenoprotein.$scaf.cDNA > ./$selenoprotein/proteina/$selenoprotein.$scaf.translate.fa
Els arxius que genera el Fastatranslate contenen 6 seqüències fasta, corresponents a les 6 possibles pautes de lectura (3 directes i 3 reverse). En el nostre cas escollíem de forma automatitzada el primer marc de lectura de la cadena directe, d'acord amb l'alineament resultant del exonerate. Ara, el T-coffee podrà alinear la nostra proteïna predita amb la proteïna original de la query.
Genewise
Amb el programa genewise generem una nova anotació del gen. La seva funció és la mateixa que l'Exonarate, tot i així, al utlitzar diferents algoritmes els resultats poden variar. Es fa servir la següent comanda, tenint en compte que podem afegir -trep si analitzem una seqüència reverse:
genewise -pep -pretty -cdna -gff ./$selenoprotein/canviXU.fa ./$selenoprotein/fastasubseq/$selenoprotein.$scaf.subseq.fa > ./$selenoprotein/genewise/$selenoprotein.$scaf.genewise.gff
Genewise ens genera de forma automàtica la seqüència peptídica. Per a comparar aquesta seqüència amb la query mitjançant t-coffee, vam utilitzar i modificar el programa GFFtoFA.pl creat pel grup C4 del curs 2011-2012 (ens van concedir el permís per fer-lo servir).
Tornar a dalt
Alineament de la proteïna: T-coffee
Una vegada hem obtingut les seqüències de les proteïnes (tant de d'Exonerate com de Genewise) utilitzem el T-coffee per tal d'alinear la proteïna obtinguda amb la query inicial. D'aquesta manera podem veure si els residus d'aminoàcids de la proteïna inicial i la predita es corresponen, i fins i tot inferir homologia.
t_coffee ./Llibreria_selenoproteins_$specie/${selenoprotein}.fa ./$selenoprotein/proteina/${selenoprotein}.${scaf}.translate.fa > ./$selenoprotein/t_coffee/$selenoprotein.$scaf.aln #Per Exonerate
t_coffee ./Llibreria_selenoproteins_$specie/$selenoprotein.fa ./$selenoprotein/genewise_proteina/$selenoprotein.$scaf.genewise.fa #Per Genewise
Tornar a dalt
Cerca d'elements SECIs: SECISerch
Un cop finalitzada la cerca de selenoproteïnes, procedim amb la cerca d'elements SECIs, capaç de reclutar el factor d'elongació eFESec. Per això vam crear el programa Secis.bash, que realitza la busqueda gracies al programa SECISearch.pl disponible a /cursos/BI/bin. Utilitza la comanda:
./SECISearch.pl < ./$selenoprotein/fastasubseq/${selenoprotein}.${scaf}.subseq.fa > ./$selenoprotein/secis/${selenoprotein}.${scaf}.html
Tornar a dalt
tRNAscan-SE
Per a completar la cerca de la maquinària de síntesi de selenoproteïnes, hem volgut buscat si hi ha els tRNAs de selenocisteïna en l'organisme estudiat. Tot i així, degut a la complicada instal·lació no ha estat possible. En tot cas, la comanda a utilitzar és:
./tRNAscan-SE /cursos/BI/genomes/project_2013/Chrysemys_picta_bellii/genome.fa > /homes/users/UXXXXX/Treball_BI/tRNAscan_output.out
Tornar a dalt
Blastp contra el conjunt no redundant de la base de dades NCBI
Podem comprovar si la proteina que hem obtingut té homòlegs en altres espècies mitjançant una cerca Blastp contra el conjunt no-redundant de totes les proteines disponibles a NCBI. Es tracta d'una eina molt potent ja que si trobem selenoproteïnes homòlogues a la nostra seqüència predita, tant per exonerate com per genewise, en altres espècies, és molt indicatiu de que veritablement és una selenoproteina (sempre i quan tingui l'aminoàcid selenocisteïna). Utilitza el software netblast i per tal d'executar-lo ho fem d'aquesta manera:
export PATH=/cursos/BI/bin/netblast/bin:$PATH
export PATH=/cursos/BI/bin/ncbiblast/bin:$PATH
cp /cursos/BI/bin/ncbiblast/.ncbirc ~/
blastall -p blastp -i ./$selenoprotein/proteina/$selenoprotein.$scaf.translate.fa -d /cursos/BI/soft/selenoprofiles_2_installation/libraries/nr -o ./$selenoprotein/NCBI/$selenoprotein.$scaf.ncbi
Tornar a dalt
Selenoprofiles
Per a verificar si el nostre anàlisi ha estat correcte hem emprat el programa Selenoprofiles. Aquest programa ha estat creat per Marco Mariotti del CRG (per a més informació podeu visitar la pàgina web http://big.crg.cat/services/selenoprofiles). Selenoprofiles inclou psi-tblastn, exonerate, genewise i diferents filtrats per a tal d'annotar i predir selenoproteïnes en diferents genomes. Aquest programa es a disposició dels estudiants de Bioinformàtica al directori /cursos/BI/bin. Es tracta d'un programa molt complexe i per això triga unes hores a mostrar resultats. Per tal de poder-lo executar tot seguit, sense que es talli, el vam enviar pel sistema de cues amb la comanda següent i les PATHs corresponents:
$ export PATH=/cursos/BI/bin/ncbiblast/bin:$PATH # pel NCBI Blast
$ cp /cursos/BI/bin/ncbiblast/.ncbirc ~/ # pel NCBI Blast
$ export PATH=/cursos/BI/soft/exonerate/i386/bin:$PATH # per l'exonerate
$ export PATH=/cursos/BI/bin:$PATH # pel GeneWise, el fastaseqfromGFF.pl i el t_coffee
$ export WISECONFIGDIR=/cursos/BI/soft/wise-2.2.0/wisecfg # pel GeneWise
nohup Selenoprofiles ~/Treball_BI/selenoprofiles -t /cursos/BI/genomes/project_2013/Chrysemys_picta_bellii/genome.fa -s "Chrysemys_picta_bellii" -p eukaryotic -temp ~/temp
Tornar a dalt
Anàlisi
En l'esquema següent podeu visualitzar l'obtenció de dades pas a pas. En verd podeu observar els programes, en blau les carpetes, en requadres amb contorn taronja els fitxers dins de la carpeta de cada selenoproteïna i en requadres blaus els fitxers utilitzats a l'hora de realitzar l'anàlisi.
Un cop obtingudes les dades havent corregut els programes, s'ha realitzat un procès d'anàlisi exhaustiu d'aquests. En aquest treball s'ha seleccionat, a l'hora de presentar resultats, el millor hit en un scaffold determinat. Durant la selecció, s'han tingut en compte les possibles duplicacions o bé dominis homòlags comuns entre selenoproteïnes de la mateixa família. Ha estat una feina laboriosa, ja que moltes de les selenoproeïnes predites no estaven del tot anotades (mancaven seqüències d'aquestes) i era difícil discernir entre proteïnes amb graus d'homologia elevats.
Aquelles selenoproteïnes on vam obtenir resultats incoherents o pràcticament nuls, utilitzavem el programa SelSearch.bash per analitzar aquella selenoproteïna en concret. No nomès permet evitar possibles errors amb la interacció del usuari, sinó que permet escollir diferent valors de variable: els valor significatiu del e-value en el blast, l'inici i la longitud del la subseqüècia, realitzar genewise...
Per últim, un cop decidit quina anotació era la més significativa i la millor anotada en el nostre genoma, hem realitzat una busqueda d'informació en la filogènia de les selenoproteïnes per evitar errors en la nomenclatura d'aquestes que difereien segons l'espècie de la query. En certs casos ens ha interessat realitzar alineaments múltiples amb t-coffee (Disponible en la web t-coffee) i transofrmar-ho en imatges apreciables utilitzatn Jalview.
Tornar a dalt