

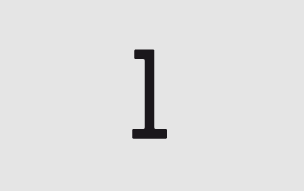

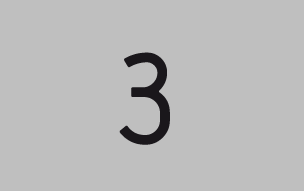





1. Obtenció dels genomes dels protists:
Els genomes dels protists analitzats han estat facilitats pel departament de Bioinformàtica de la Universitat Pompeu Fabra, a través del seu cluster de docència, accessible des de la següent ruta:
~/cursos/BI/genomes/protists/2011
Aquests catorze genomes no estan completament ensamblats, i s'organitzen en regions o contigs de longitud variable.
2. Obtenció de les seqüències queries de Selenoproteïnes:
Query significa literalment dubte, pregunta o qüestió. En el nostre context s'entén com la proteïna o regió proteica coneguda que ens permetrà establir la seva possible relació amb el genoma en estudi.
Les seqüències de selenoproteïnes conegudes utilitzades han estat obtingudes majoritàriament de la base de dades SelenoDB, de la qual, entre les múltiples opcions que ens ofereix, hem seleccionat la cadena polipeptídica. D'altra banda, en aquells casos en què aquesta font era insuficient, hem recorregut a altres fonts com NCBI: protein i a UniProt. En tots els casos, les seqüències s'han emmagatzemat en fitxers FASTA.
Per tal de poder estudiar un marc evolutiu més ampli, hem emprat, quan ha estat possible, queries procedents d'organismes molt distanciats filogenèticament.
Durant la realització de l'estudi, les queries han estat designades en funció de la seva família, subfamília i espècie de la que provenen segons el següent patró:
$ familia_especie_subfamilia.aa.fa
Tornar a l'inici
3. Alineament de seqüències, tblastn:
BLAST (Basic Local Alignment Search Tool) és un algoritme que permet comparar seqüències biològiques, ja siguin aminoacídiques o nucleotídiques. De les múltiples opcions que ens ofereix BLAST, la que més s'ajusta a les nostres necessitats és TBLASTN.
TBLASTN permet comparar una seqüència proteica queries amb una de nucleotídica inclosa en un fitxer o base de dades i traduïda en totes les possibles pautes de lectura.
Per tal d'utilitzar aquest software, l'exportem al nostre shell mitjançant la següent comanda:
$ export PATH=/cursos/BI/bin/ncbiblast/bin:$PATH
$ cp /cursos/BI/bin/ncbiblast/.ncbirc ~/
A continuació, cal formatejar la base de dades de genomes esmentada anteriorment amb la següent comanda:
$ formatdb -i /cursos/BI/genomes/protists/2011/organisme/genome.fa -p F -n genome_organisme.fa
Llavors, executem el TBLASTN:
$ blastall -p tblastn -i query.fa -d basededades.fa -o fitxerdesortida
Que, en el nostre cas, la comanda esdevé:
$ blastall -p tblastn -i familia_especie_subfamilia.aa.fa -d genome_organisme.fa -o tblastn_familia_especie-vs-genoma.fa
Aquest programari presenta nombroses opcions o arguments per tal d'introduir canvis en l'anàlisi de les dades i el contingut dels resultats obtinguts, d'entre els quals ens són d'especial utilitat:
-m 9: tabula els resultats i en descriu la informació més rellevant (identificador del contig, longitud de l'alineament, inici i final, e-value, etc).
-F F: autoritza l'alineament de les Low Complexity Regions i permet observar amb més detall els alineaments.
- e: filtra els resultats i només mostra aquells que són d'e-value inferior a un valor determinat per l'usuari, que en nostre cas vam establir en 0,001.
Filtratge dels resultats de TBLASTN
La quantitat de dades processades genera, conseqüentment, un gran volum de resultats. D'aquests, però, nomès seleccionem per al seu estudi, aquells que presenten un bon alineament entre la query emprada i el genoma en qüestió.
El propi TBLASTN ens considera la qualitat de l'alineament de manera inversament proporcional a un valor anomenat e-value. Aquest ès, doncs, el primer filtre que utilitzem per escollir aquells arxius que posteriorment analitzarem. Per la naturalesa del nostre estudi, tambè són prioritaris aquells resultats en els quals la selenocisteïna (U) de la query alinea amb un codó STOP (*) o amb una cisteïna (C). Així mateix, tenen un especial valor aquells hits que, de manera simultània en diferents alineaments, coincideixen en el mateix contig o fragment de genoma.
Tornar a l'inici
4. Anotació del genoma:
-Exonerate
Exonerate és una eina informàtica que permet comparar seqüències utilitzant nombrosos models d'alineament. En el nostre cas ens permet, per banda, extreure la regió genòmica d'interès i per l'altra, l'anotació del genoma per tal de predir la possible estructura exònica de la proteïna problema. Per tal de fer servir aquest programari, l'exportem amb la següent comanda:
$ export PATH=/cursos/BI/bin/exonerate/i386/bin:$PATH
Primerament, indexem el genoma del microorganisme en contigs ordenats:
$ fastaindex /cursos/BI/genomes/protists/2011/organisme/genome.fa genome_organisme_index.fa
Seguidament, extraiem la seqüència nucleotídica delimitada pel contig que conté la possible selenoproteïna mitjançant fastafetch, i, a partir d'aquesta, la regió d'interès amb fastasubseq. Aquests dos programes acompanyen l'exonerate, i les dues comandes corresponents són:
$ fastafetch /cursos/BI/genomes/protists/2011/organisme/genome.fa genome_organisme_index.fa 'identificadordelcontig' > genome_familia_organisme_fetch.fa
$ fastasubseq genome_organisme_fetch.fa start length > genome_familia_organisme_subseq.fa
En aquest últim cas, per assegurar-nos que “abarquem” tota la seqüència de la possible proteïna, ampliem ambdós marges del hit 10000 nucleòtids.
Un cop hem extret la regió genòmica d'interès, utilitzem exonerate per tal de predir l'estructura exònica de la nostra possible proteïna a través de la següent ordre al shell:
$ exonerate -m p2g –showtargetgff –exhaustive yes -q familia_organisme_subfamilia.aa.fa -t genome_familia_organisme_subseq.fa | egrep -w exon > genome_familia_organisme_exonerate.gff
En aquesta comanda, incloem, com es pot comprovar, alguns dels arguments que ens ofereix exonerate:
-m p2g: indica que comparem un model proteic contra un de nucleotídic.
-- exhaustive yes: incrementa l'exhaustivitat de la nostra cerca
egrep -w exon: aquest pipe ens permet descartar aquelles línies del fitxer que no contenen exons.
El format del fitxer que obtenim amb l'exonerate és .gff, i per tant, cal transformar-lo a FASTA per poder continuar el nostre estudi. El programa que ens permet aquesta conversió és un Perl, que ens extraurà, doncs, el cDNA de la proteïna:
$ fastaseqfromGFF.pl genome_familia_organisme_subseq.fa genome_familia_organisme_exonerate.gff > cDNA_familia_organisme.fa
Així doncs, obtenim un fitxer amb la seqüència de nucleòtids que el programa fastatranslate ens tradueix a aminoàcids. A la comanda hi afegim l'opció -F 1 per tal que ens mostri la seqüència resultant de la millor pauta de lectura:
$ fastatranslate -F 1 cDNA_familia_organisme.fa > prot_familia_organisme.fa
-GeneWise
Tot i que preferencialment utilitzem exonerate, existeix una alternativa per a l'anotació del genoma. Aquesta és un programa anomenat GeneWise que s'aplica sobre l'arxiu obtingut del fastasubseq, obtingut del procediment explicat anteriorment. Els permisos que ens permeten utilitzar-lo i la comanda corresponent són:
$ export PATH=/cursos/BI/bin:$PATH
$ export WISECONFIGDIR=/cursos/BI/soft/wise-2.2.0/wisecfg
$ genewise -pep -pretty -cdna -gff -both familia_organisme_subfamilia.aa.fa genome_familia_organisme_subseq.fa > genome_familia_organisme_genewise.gff
GeneWise, a diferència de l'exonerate, és sensible a la direccionalitat de l'alineament i és per això que utilitzem l'opció -both.
Tornar a l'inici
5. Comparació de les seqüències aminoacídiques, TCOFFEE:
Un cop hem obtingut la seqüència de la nostra possible proteïna, l'alineem amb les queries utilitzades inicialment (les quals hem agrupat en un fitxer Fasta) amb el programa TCOFFEE:
$ export PATH=/cursos/BI/bin:$PATH
$ t_coffee prot_familia_organisme.fa queries_familia.fa > tcoffee_familia_organisme.fa
L'alineament obtingut, que se'ns mostra tant en format Fasta com html, ens permet una visualització de la qualitat de l'alineament i, alhora, de la conservació ineterspecífica de la nostra regió d'interès.
Tornar a l'inici
6. Cerca de proteïnes homòloques, BLASTPs:
BLASTP estableix una comparació entre la proteïna obtinguda i la base de dades de GenBank amb l'objectiu de trobar homologies amb altres espècies que no hem estudiat. Així, podem demostrar la conservació de determinats dominis clau en la seqüència d'aminoàcids obtinguda.
Aquesta funció ve donada com a recurs web: BLASTP
Tornar a l'inici
7. Estudi dels elements SECIS:
Amb l'objectiu d'identificar la presència d'elements SECIS, utilitzem la pàgina web SeciSearch, a la qual introdïm el fitxer subsequence.fa obtingut en els passos anteriors. La cerca es pot dur a terme en les dues direccions del DNA i a diferents nivells d'exigència.
Inciem la nostra cerca amb el màxim d'exigència, strict, i en cas de no obtenir resultats, relaxem els criteris de l'anàlisi amb les opcions canonical i non-canonical malgrat el risc d'obtenir falsos positius. Així mateix, cal considerar que el cove score del resultat obtingut hauria de ser, idealment, igual o superior a quinze.
Tornar a l'inici
8. Automatització del procés:
Degut al gran volum de dades inicials, i per facilitar la nostra tasca, hem ideat un algoritme en suport Bash que ens automatitza l'alineament amb TBLASTN i, a més, l'organització dels resultats en un arbre de directoris estructurats.
Tornar a l'inici