| Introduction | Goals | Methodology | Programs | Results | Conclusions | References | Authors | Acknowledgements |
RESULTS
The exhaustive analysis of results detailed above is available in Conclusions. |
||||||||||||||||||||||||||||||
Analysis of algorithm complexity and computational cost of brute-force pattern matching algorithm versus Knuth-Morris-Pratt algorithm In order to determine the computational achievement of Knuth-Morris-Pratt algorithm comparing with force-brute pattern matching algorithm, we implemented two programs for each one, which function is generating all the possible patterns of a determined mRNA and verifying if those patterns are unique or not in the same mRNA, with the correspondence algorithm. |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||
Reproduction of Mus musculus mRNA results by Knuth-Morris-Pratt algorithm In order to corroborate the results obtained by Dr. Robert Castelo, Mus musculus EPO gene, we implemented our program of unique subsequences search (explained in Programs) based on Knuth-Morris-Pratt algorithm, but only in mouse chromosome 5 to save time, because EPO gene locates in this chromosome.
Obtained results are that the following subsequences are unique:
|
||||||||||||||||||||||||||||||
CGACAGTCGAGTTC
AGTGGTCTACGTAG
GGGTCTACGCCAAC
|
||||||||||||||||||||||||||||||
Moreover, the program also informs about the shift of these patterns in the chromosome, a very imporant fact for using the results to its application in relation to gene doping, and also in those cases that found subsequences locate in two different exons separated by intron. The found shift for each subsequence is the next one: |
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||
As our program has a modifyng the length automatic strategy (in order to find the minimum unique subsequences) and we only checked in chromosome 5, we got also minor length subsequences than 14 nucleotides for the first and third subsequences, that if we had checked in whole genome, subsequences would have not been uniques.
For the first subsequence, the program finds one subsequence of 12 nucleotides: CGACAGTCGAGT, with a shift of 136.403.438. And for the third subsequence, it finds a subsequence of 11 nucleotides: GGGTCTACGCC, with a shift of 136.401.415. If we compare the shift of these smaller sebsequences with the shift of the subsequences they come from, it does not agree with initial position. This is because all the subsequences are found in the negative strand. Considering this fact, the shift coincides.
|
||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||
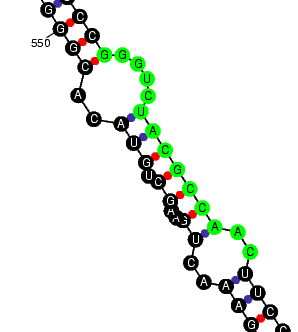
Prediction of EPO mRNA secondary structure To elucidate if the found unique subsequences are located in a exposed region of the mouse EPO mRNA we have used the mFOLD program. This program predicts the folding package of a mRNA or DNA sequence. The results are the next ones:
|
||||||||||||||||||||||||||||||
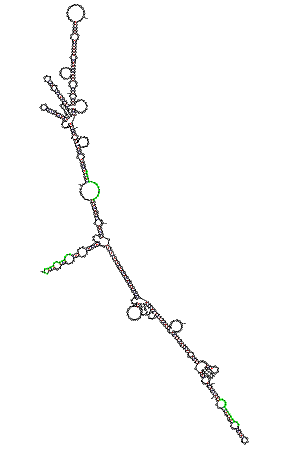
Click on the image to get the postscript that allows you to blow up the mRNA structure.
The three unique subsequences are marked in green.
|
||||||||||||||||||||||||||||||
Now, the three unique subsequences are enlarged separately: |
||||||||||||||||||||||||||||||

 | ||||||||||||||||||||||||||||||