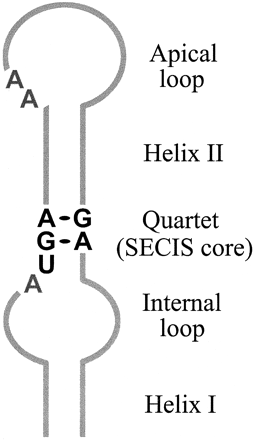
The UGA codon was thought to have a unique meaning as the STOP signal of the genetic code. But after discovery of selenoproteins another meaning has been attributed to this codon as soon as it can incorporate a novel aminoacid called “selenocysteine”(Sec). This newly found aminoacid is not more than a cysteine having selenium instead of sulphur. The condition for selenocysteine to be incorporated is having a Sec insertion sequence (SECIS) in the near 3’ untranslated region (UTR). This sequence forms a loop that enables recruitment of several factors and enzymes that permit Sec incorporation to the nascent polypeptide chain.
{kind=link}
The functional importance of these proteins remains poorly understood, but it seems that selenocysteine is often located in the enzyme active-sites and is usually essential for its activity. This fact confers an enhanced value to the search and characterization of these novel proteins not only in humans but also in other organisms. 25 selenoproteins have been described so far for humans but we still don’t know if similar proteins are present in insects whom genomes sequentiation is in advanced phase if not completed.
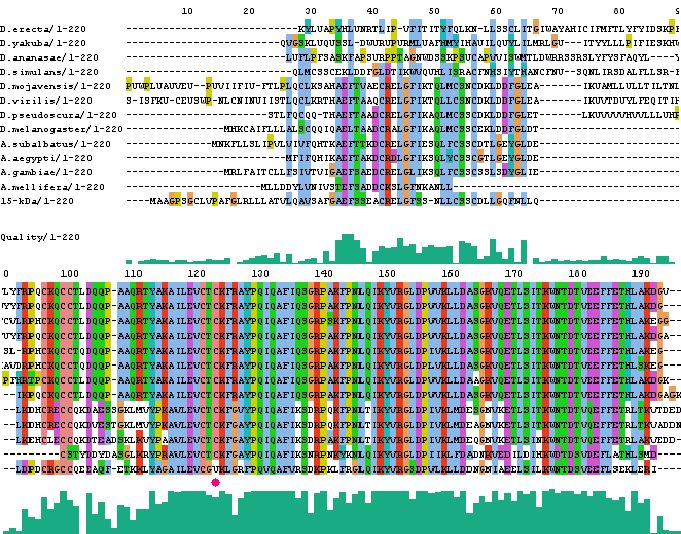
In this project we report the presence of proteins similar to those found in humans in some organisms belonging to the family arthropoda. We have focused our study in organisms having the genome sequenced enough such as Anopheles gambiae, Apis mellifera, Drosophyla pseudoscura or others such as Drosophyla melanogaster with completed genome sequentiation. Thus, we have performed similarity search (tBLASTn) followed by exonic structure analysis (exonerate), SECIS structure prediction (SeciSearch) and multiple alignment of our hits (ClustalW).
Results of BLAST (hits) showing different scores and conservation levels were submitted to exonerate to further analyse the exonic structure of our putative proteins paying special attention to location of the aminoacids matching with selenocysteine (expressed as a STOP) and its adjacencies. Considering that an in-frame TGA (STOP) signal located not at the end but between other triplets could suggest a UGA coding for selenocysteine.
SeciSearch allowed us to identify putative SECIS elements in order to emphasize the information obtained by exonerate analysis or to discard hits that even having TGA inside the exon lack the necessary SECIS to be interpreted as possible selenoproteins.
Finally, the multiple alignment was done aimed at knowing the conservation level among our putative proteins, considering that a high conservation suggests that aminoacids are part of a functional protein and keep restrictive to mutations that would possibly affect worsening the activity.
2. Methods
SELENOPROTEINS AND ORGANISMS
For this project, we have used the 25 selenoproteins described in mammals so far.
Among these 25 proteins only three have been found in Drosophyla melanogaster also known as "fruitfly", but the presence of homologues in other insect species is still unknown.
The aim of this study is to discover if homologues of the mammalian 25 seleproteins are present in insects, and with this purpose we have performed tblastn similarity search of the 25 selenoproteins against several insect genomes, in order to dilucidate their general distribution.
In the beginning we were suposed to perform the study with three insect genomes belonging to Drosophyla simulans, Anopheles gambiae and Appis mellifera. Even though, we considered much more interesting to extend our study to more genomes, mainly because the results would be much more emphazised if they were supported by more organisms.
Thus, we submitted all the species belonging to the family arthropoda, having a partially or completelly sequenced genome, to similarity search.
Especies from genus Drosophila:
D.melanogaster [C,G,P,D]
D.pseudoobscura [C,G,P,D
D.ananassae [S,D]
D.erecta [S,D]
D.yakuba [S,D]
D.mojavensis [S,D]
D.simulans [S,D]
D.virilis [S,D]
D.grimshawi [D]
D.persimilis [D]
D.willistoni [D]
D.sechellia [-]
C = published Chromosomes (may be partial);
S = preliminary assembly Scaffolds;
G = genome Genes;
P = genome Proteins; - = no data yet
D = Databank sequences (NT) from GenBank/EMBL, RefSeq; Databank proteins (AA) from UniProt, RefSeqp, GenPept;
Other species:
Apis mellifera
Anopheles gambiae
Armigeres subalbatus
Aedes aegypti
Glossina morsitans morsitans
Ixodes ricinus
Venturia canescens
Musca domestica
Bombyx mori
PROGRAMMES
SIMILARITY SEARCH
TBLASTN
Compares a protein query sequence against a nucleotide sequence database dynamically translated in all six reading frames (both strands) using the BLAST algorithm. The BLAST seeks local as opposed to global alignments and is therefore able to detect relationships among sequences which share only isolated regions of similarity (Altschul et al., 1990).
In our work, NCBI tblastn has been used with two differents databases; the insects genomes NCBI database and Drosophila Species Genomes, a service of FlyBase and Genome Sequencing Centers. This service provides a preview of drosophila genome data, with genome maps and blast sequence search for this species. It includes prepublication, as well as published, Drosophila genomes from collaborating NGHRI-funded Genome Sequencing Centers.
To run tblastn program, we have paid special attention to some parameters,assuming that they were the most significant and informative for our purpose.
Expect: The statistically signifant expectation value. If the statistical significance ascribed to a match is greater than the E value, the match will not be reported. As soon as we were comparing distant species, we avoided the established E-value to be stringent, in order to obtain more candidate hits. We used the default value(=1), but we assumed that only E-values below 0.5 would be taken into account.
Substitution matrix: As it is known, this matrix assigns a score for aligning any possible pair of residues based on the theory of amino acid substitutions. In general, different substitution matrices are tailored to detecting similarities among sequences that are diverged by differing degrees. A single matrix may nevertheless be reasonably efficient over a relatively broad range of evolutionary change. Experimentation has shown that the BLOSUM-62 matrix is among the best for detecting most weak protein similarities. For particularly long and weak alignments, the BLOSUM-45 matrix may prove superior.
In our study, we established the BLOSUM45 matrix for our similarity search which is thought to be the most appropriate for the low evolutionary kinship between our subject and query.
GENE PREDICTION
First of all the use of Geneid for gene prediction was planned. Nevertheless, this turned to be uneffective in our case as soon as our subject insect genomes were not available.Thus, we realised that programs not depending on intrinsec databases were needed.Two options were discussed; Exonerate and Genewise; after following our tutor's advices, the first one was chosen.
EXONERATE protein2genome 0.8.2
Exonerate is a general tool for sequence comparison that uses the C4 dynamic programming library. It can produce either gapped or ungapped alignments according to a variety of different alignment models. Alignments generated using these heuristics will represent a valid path through the alignment model, yet (unlike the exhaustive alignments), the results are not guaranteed to be optimal.
We have used exonerate to check the exonic structure and intron moddeling of our hits. The aim was to observe whether the -STOP codon-, candidate to be selenocystein, was located in-frame or not. Considering that a positive answer to this question would further verify the posibility of having discovered a selenoprotein.
Anyway, the candidates not showing any result after being submitted to exonerate have not been discarded. We have considered that the query ( human selenoprotein) could be so distant that the exonerate would not accept the target DNA to contain any exonic structure ( that meens protein),related to the query. In order to solve this doubt we thought that the information given by the later multiple alignment could be clarifying, thinking that the insect protein sequences could point out to be very conserved among insects ( suggesting that they are part of proteins) , even not giving any results in exonerate analysis.
We performed a gapped alignment exonerate analysis with the following parameters:
SEQUENCE INPUT OPTIONS
Pairwise comparisons will be performed between all query sequences and all target sequences. In our case, human selenoprotein sequences in fasta format are used as the query sequences(-q), and insect genomic sequences also in fasta(-t).
-q | --query
Human selenoprotein sequence in a FASTA format file.
-t | --target
The insect DNA sequence also in a FASTA format file.
GAPPED ALIGNMENT OPTIONS
-m | --model
This model allows alignment of a protein sequence to genomic DNA. This is similar to the protein2dna model, with the addition of modelling of introns and intron phases. This model is simliar to those used by genewise.
--showtargetgff
Report GFF output for features on the target sequence. We show our exonerate results in a GFF file showing the exonic structurend intron moddeling.
MULTIPLE ALIGNMENT
FASTACHUNK
Fastachunk is a tool that enables us to cut the desired DNA fragment from a file containig a sequence. We used it when the stop"Sec" matching aminoacids hadn't been found with tblastn in order to find out the upstrean or downstrean aminoacids sequence with a later " Translate tool" program. Thus, we could realize if the stop wasn't really matching with Sec or Cys. Nevertheless, we thought that we also could find a stop codon or Cys in our subject matching with the human protein's stop,but not shown due to the penalization attributed by the program to the STOP.
Another reason why Fastachunk was used, is because the fragments added could presumably show conservation in the later multiple alignment.This could mean that even not matching with the human selenoprotein (for being distant species), they have enough conservation among insects to be considered as parts of the protein constituents.
TRANSLATE TOOL
Translate is a tool which allows the translation of a nucleotide (DNA/RNA) sequence to a protein sequence in the all six reading frames.This allowed us to recover the protein sequence of the DNA fragments submitted to Fastachunk.Thus, we could clarify the questions above, comparing the protein sequence to that obtained in tblastn and also submit them to multiple alignment.
CLUSTALW
Clustal W is a general purpose multiple sequence alignment program for DNA or proteins.It produces biologically meaningful multiple sequence alignments of divergent sequences. It calculates the best match for the selected sequences, and lines them up so that the identities, similarities and differences can be seen. Evolutionary relationships can be seen via viewing Cladograms or Phylograms.
This tool has enabled us to clarify the distribution of the selenoproteins in different organisms and check the conservation level of the same putative protein among them.
When running the clustalW programme, all the protein sequences matching with the human specific selenoprotein were submitted together in fasta format. The sequences were enlarged compared to the specific fragment matching with the human selenoprotein, a work that was performed using FASTACHUNK program.
All the clustalW parameters remained as they were given by default for our multiple alignment.
3. Results
Results are given for each protein in the following order:
- tblastn hits: blast reference E-value Identities
- Exonerate output in GFF format or tblastn output for the cases with no results in exonerate.
- Secis elements: reference sequence score picture
- ClustalW output and Jalview edited multiple alignment.
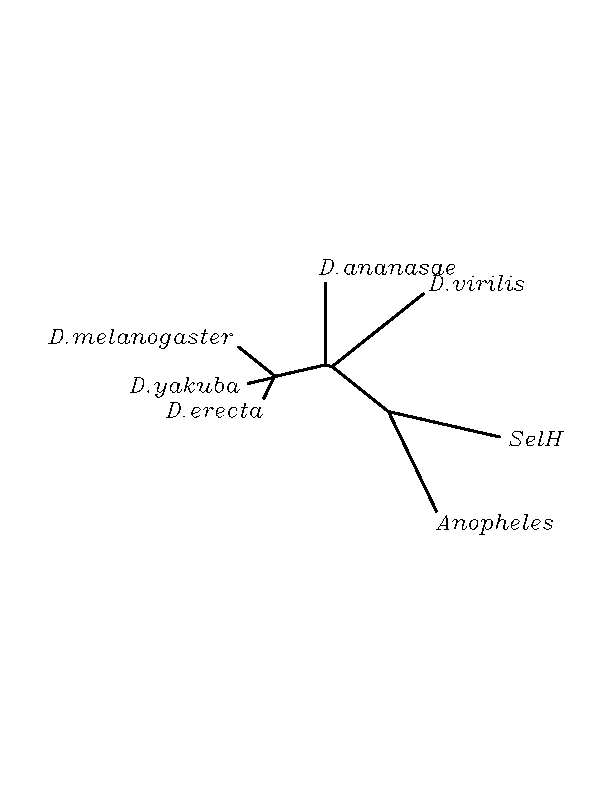
SelH
6 hits are given by the tblastn similarity search, but only four of them were accepted to run exonerate analysis. The four sequences submitted to this analysis showed to have their "Stop codon" outside the coding regions according to the exonerate program. Even though, SECIS structure analysis reinforced the hypothesis of classifying them as selenoproteins showing very well scored SECIS structures within the six sequences, even the two sequences that were not admitted to run exonerate.
tblastn
>gnl|Dana-agencourt-040714-asm|contig_3129 1e-07 37% >gnl|Dyak-washu-assembly_040407|Contig124.5 2e-07 36% >0gnl|Dere-agencourt-run1028-asm|contig_8152 4e-07 35% >gnl|Dvir-agencourt-run1029-asm|contig_2170 0.044 25% >gi|27645578|emb|BX072297.1|CNS09RY5 Single read from an extremity of a full-length cDNA clone made from Anopheles gambiae total adult females. 2e-14 37% >gi|21430733|gb|AY119185.1| Drosophila melanogaster SD09114 full insert cDNA 4e-10 33%
Gene prediction
If you want to check all the exonerate outputs in gff format, click here.
SECIS elements
gnl|Dana-agencourt-040714-asm|contig 3129: 8621 8720
AUAGAAAUCU CCGCCAG CAAUAGGCCUU AUGAA GUGUGAUCGGCC AA UCCUCUCCAGGA ACCGACCACAC UGAA CCACAAUUU UUGUUUG CCUUUGUCUG
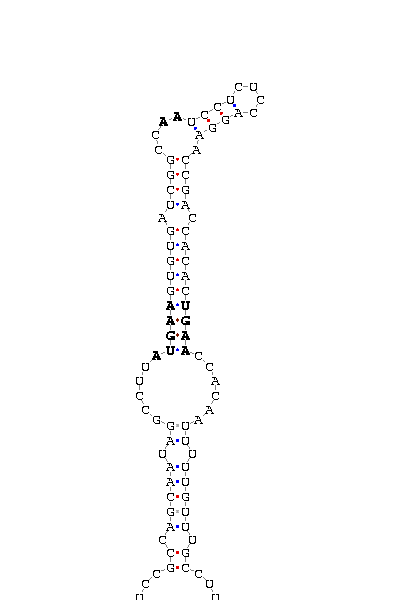
COVE score: 27.13 (Recommended threshold for COVE: 15)
gnl|Dere-agencourt-run1028-asm|contig 8152: 905 807 (SECIS on complementary strand)
AACAAACCUU UCUACAA UAGAGCCCC AUGAU CGAUGAUUGGC AA AUCCUCUCGAGGA ACCAAUUAUUG AGAA CCUUUUUGCC UUUGUUG AUUGUAUAAU
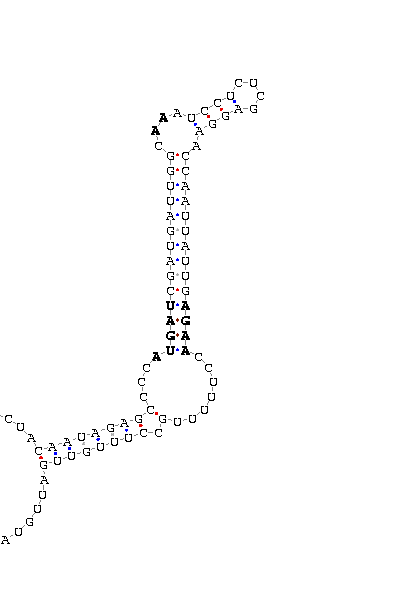
COVE score: 21.21 (Recommended threshold for COVE: 15)
gnl|Dvir-agencourt-run1029-asm|contig 2170: 20539 20639
GCGUCGAUAG UUUUUCA GCAAUAGCGCCAU AUGAU CAUUAAACGGU AA AACCCUAACGGGA GCCGUUCACUG UGAA ACUUUUGCC UGUAAG CUGAAGAAUU
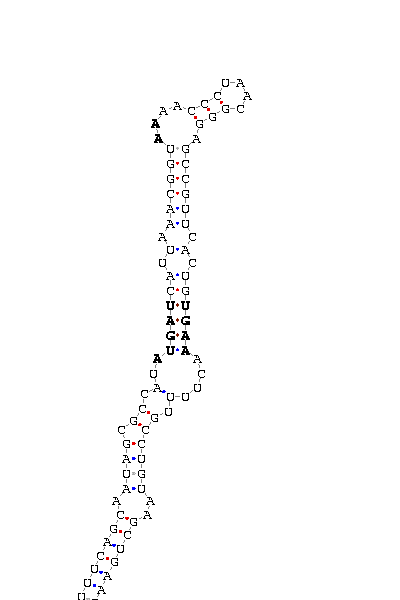
COVE score: 20.43 (Recommended threshold for COVE: 15)
gnl|Dyak-washu-assembly 040407|Contig124.5: 4158 4059 (SECIS on complementary strand)
AAACGAAUCU UUCAGCA AUAGAGCCUU AUGAU CGAUGAUUGGC AA AUCCUUUCGAGGA ACUUAUCAUUG AGAA CCCUUUUGCC UUUGUUG AUUGCUUAGU
COVE score: 18.32 (Recommended threshold for COVE: 15)
>gi|27645578|emb|BX072297.1|CNS09RY5 Single read from an extremity of a full-length cDNA clone made from Anopheles gambiae total adult females. 5-PRIME end of clone FK0AAD1AF11 of strain 6-9 of Anopheles gambiae (African malaria mosquito): 640 734
CUUUAAUUUC UUCGAAU CGCCAUGCCGC AUGAC GAAGCCUGAGC AA ACCCCACGUGGGA CUCGAGCCUU UGAA GCUCU GCUGGCG UUUCGGUAGA
COVE score: 24.71 (Recommended threshold for COVE: 15)
>gi|21430733|gb|AY119185.1| Drosophila melanogaster SD09114 full insert cDNA: 816 917
CAAAACAAAG CGUUCAG CAAUAGAGCCCU AUGAU CGAUGAUUGGC AA AUCCUCUCGAGGA ACCGAUCGUUG AGAA CCCCUUUGCC UUUGUUG AUCGCUCAAU
COVE score: 21.48 (Recommended threshold for COVE: 15)
Multiple alignment
If you want to see the ClustalW alignment, click here.
Unrooted N-J tree
SPS2
SPS2 human selenoprotein matched with high homology in many of the insect genomes submitted to tblastn. Here we report the discovery of two new selenoprotein candidates belonging to the species: D.yakuba and D.simulans. All the other insect species were found to have an "Arg" aminoacid at the position of the "Sec" of humans.
tblastn
>gnl|Dvir-agencourt-run1029-asm|contig_641 e-112 60% >gnl|Dmov-agencourt-run0811-asm|contig_3459 e-112 60% >gnl|Dana-agencourt-040714-asm|contig_1299 e-111 60% >gnl|Dere-agencourt-run1028-asm|contig_125 e-111 59% >gnl|Dyak-washu-assembly_040407|Contig11.8 e-111 59% >3 type=chromosome; loc=3:1..19738957; ID=3; release=r1.03; species=dpse e-110 59% >gnl|Dana-agencourt-040714-asm|contig_30531 2e-97 58% >XR_group8 type=chromosome; loc=XR_group8:1..9190824; ID=XR_group8; release=r1.03; species=dpse 2e-73 43% >4_group4 type=chromosome; loc=4_group4:1..6604331; ID=4_group4;release=r1.03; species=dpse 2e-68 42% >XL_group1e type=chromosome; loc=XL_group1e:1..12499574; ID=XL_group1e; release=r1.03; species=dpse 2e-54 38% >gnl|Dyak-washu-assembly_040407|Contig8.19 7e-52 38% >4_group3 type=chromosome; loc=4_group3:1..11635473; ID=4_group3;release=r1.03; species=dpse 2e-52 36% >gnl|Dana-agencourt-040714-asm|contig_1089 3e-51 39% >gnl|Dsim-washu-w501-asm|Contig27.138 3e-51 39% >gnl|Dvir-agencourt-run1029-asm|contig_2233 6e-31 44% >gnl|Dmov-agencourt-run0811-asm|contig_181 0.060 50% >gnl|Dsim-washu-w501-asm|Contig18.273 3e-06 41% >gi|24653603|ref|NM_166046.1| Drosophila melanogaster CG8553-PB, isoform B (SelD) mRNA, complete cds e-130 60% >gi|58390119|ref|XM_317503.2| Anopheles gambiae str. PEST ENSANGP00000010100 e-126 59%
Gene prediction
If you want to check all the exonerate outputs in gff format, click here.
For the sequences that exonerate could not have been performed, tblastn outputs are shown:
>gnl|Dmov-agencourt-run0811-asm|contig_717 Length = 57670 Score = 41.8 bits (131), Expect = 0.003 Identities = 18/44 (40%), Positives = 24/44 (54%) Frame = -1 Query: 12 FQNHFEPGVYVCAKCGYELFSSRSKYAHSSPWPAFTETIHADSV 55 + H+E GVY C C +LFSS +KY WPAF + + V Sbjct: 21133 YNKHYEKGVYRCIVCHQDLFSSDTKYDSGCGWPAFNDVLDKGKV 21002 Score = 36.5 bits (112), Expect = 0.13 Identities = 21/46 (45%), Positives = 29/46 (63%) Frame = -3 Query: 59 PEHNRSEALKVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFV 104 PE R+E V C +C +GH F DGPKP + R+ I S+S++FV Sbjct: 18638 PERIRTE---VRCARCNAHMGHVF-EDGPKPTRKRYCINSASIEFV 18513 Score = 35.1 bits (107), Expect = 0.36 Identities = 18/37 (48%), Positives = 24/37 (64%) Frame = -2 Query: 68 KVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFV 104 +V C KC +GH F +DGP P + RF I S+S+ FV Sbjct: 20268 EVRCSKCSAHMGHVF-DDGPPPKHRRFCINSASIDFV 20161
>gnl|Dana-agencourt-040714-asm|contig_4797 Length = 61609 Score = 41.5 bits (130), Expect = 0.004 Identities = 18/44 (40%), Positives = 24/44 (54%) Frame = -3 Query: 12 FQNHFEPGVYVCAKCGYELFSSRSKYAHSSPWPAFTETIHADSV 55 + H+E GVY C C +LFSS +KY WPAF + + V Sbjct: 43103 YNKHYEKGVYQCIVCHQDLFSSDTKYDSGCGWPAFNDVLDKGKV 42972 Score = 40.4 bits (126), Expect = 0.009 Identities = 25/55 (45%), Positives = 34/55 (61%) Frame = -3 Query: 59 PEHNRSEALKVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFVPKGKETSAS 113 PE R+E V C +C +GH F DGPKP + R+ I S+S++FV K+ SAS Sbjct: 40394 PERIRTE---VRCARCSAHMGHVF-EDGPKPTRKRYCINSASIEFVTGEKDPSAS 40242 Score = 35.6 bits (109), Expect = 0.24 Identities = 27/70 (38%), Positives = 37/70 (52%), Gaps = 1/70 (1%) Frame = -3 Query: 36 KYAHSSPWPAFTETIHADSV-AKRPEHNRSEALKVSCGKCGNGLGHEFLNDGPKPGQSRF 94 K+ HSS TI + V +K R+E V C +C +GH F +DGP P + RF Sbjct: 42236 KFRHSSHTSITVNTIKSTVVISKTLGMVRTE---VRCSRCSAHMGHVF-DDGPPPKHRRF 42069 Query: 95 *IFSSSLKFV 104 I S+S+ FV Sbjct: 42068 CINSASIDFV 42039
>gnl|Dyak-washu-assembly_040407|Contig7.13 Length = 111046 Score = 41.5 bits (130), Expect = 0.004 Identities = 18/44 (40%), Positives = 24/44 (54%) Frame = +1 Query: 12 FQNHFEPGVYVCAKCGYELFSSRSKYAHSSPWPAFTETIHADSV 55 + H+E GVY C C +LFSS +KY WPAF + + V Sbjct: 46012 YNKHYEKGVYQCIVCHQDLFSSDTKYDSGCGWPAFNDVLDKGKV 46143 Score = 37.6 bits (116), Expect = 0.062 Identities = 23/54 (42%), Positives = 32/54 (59%) Frame = +1 Query: 59 PEHNRSEALKVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFVPKGKETSA 112 PE R+E V C +C +GH F DGPKP + R+ I S+S++FV TS+ Sbjct: 48979 PERIRTE---VRCARCNAHMGHVF-EDGPKPTRKRYCINSASIEFVNADPATSS 49128 Score = 35.3 bits (108), Expect = 0.29 Identities = 18/45 (40%), Positives = 26/45 (57%) Frame = +2 Query: 68 KVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFVPKGKETSA 112 +V C +C +GH F +DGP P + RF I S+S+ FV + A Sbjct: 47174 EVRCSRCSAHMGHVF-DDGPPPKHRRFCINSASIDFVKSATPSKA 47305
>gnl|Dvir-agencourt-run1029-asm|contig_386 Length = 191045 Score = 41.5 bits (130), Expect = 0.004 Identities = 18/44 (40%), Positives = 24/44 (54%) Frame = -1 Query: 12 FQNHFEPGVYVCAKCGYELFSSRSKYAHSSPWPAFTETIHADSV 55 + H+E GVY C C +LFSS +KY WPAF + + V Sbjct: 176732 YNKHYEKGVYQCIVCHQDLFSSDTKYDSGCGWPAFNDVLDKGKV 176601 Score = 36.5 bits (112), Expect = 0.13 Identities = 21/46 (45%), Positives = 29/46 (63%) Frame = -3 Query: 59 PEHNRSEALKVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFV 104 PE R+E V C +C +GH F DGPKP + R+ I S+S++FV Sbjct: 174108 PERIRTE---VRCARCNAHMGHVF-EDGPKPTRKRYCINSASIEFV 173983 Score = 35.6 bits (109), Expect = 0.24 Identities = 18/39 (46%), Positives = 25/39 (64%) Frame = -3 Query: 68 KVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFVPK 106 +V C KC +GH F +DGP P + RF I S+S+ FV + Sbjct: 175752 EVRCSKCSAHMGHVF-DDGPPPKHRRFCINSASIDFVKR 175639
>2 type=chromosome; loc=2:1..30711475; ID=2; release=r1.03; species=dpse Length = 30711475 Score = 41.5 bits (130), Expect = 0.004 BLAST HIT on Genome Map Identities = 18/44 (40%), Positives = 24/44 (54%) Frame = -3 Query: 12 FQNHFEPGVYVCAKCGYELFSSRSKYAHSSPWPAFTETIHADSV 55 + H+E GVY C C +LFSS +KY WPAF + + V Sbjct: 28012277 YNKHYEKGVYQCIVCHQDLFSSDTKYDSGCGWPAFNDVLDKGKV 28012146 Score = 36.5 bits (112), Expect = 0.13 BLAST HIT on Genome Map Identities = 21/46 (45%), Positives = 29/46 (63%) Frame = -1 Query: 59 PEHNRSEALKVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFV 104 PE R+E V C +C +GH F DGPKP + R+ I S+S++FV Sbjct: 28009705 PERIRTE---VRCARCNAHMGHVF-EDGPKPTRKRYCINSASIEFV 28009580 Score = 35.9 bits (110), Expect = 0.20 BLAST HIT on Genome Map Identities = 22/46 (47%), Positives = 29/46 (63%) Frame = -2 Query: 68 KVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFVPKGKETSAS 113 +V C KC +GH F +DGP P + RF I S+S+ FV K T+AS Sbjct: 28011345 EVRCSKCSAHMGHVF-DDGPPPKHHRFCINSASIDFV-KSAPTAAS 28011214
>gnl|Dere-agencourt-run1028-asm|contig_1017 Length = 31091 Score = 41.5 bits (130), Expect = 0.004 Identities = 18/44 (40%), Positives = 24/44 (54%) Frame = +3 Query: 12 FQNHFEPGVYVCAKCGYELFSSRSKYAHSSPWPAFTETIHADSV 55 + H+E GVY C C +LFSS +KY WPAF + + V Sbjct: 19356 YNKHYEKGVYQCIVCHQDLFSSDTKYDSGCGWPAFNDVLDKGKV 19487 Score = 37.6 bits (116), Expect = 0.062 Identities = 23/54 (42%), Positives = 32/54 (59%) Frame = +2 Query: 59 PEHNRSEALKVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFVPKGKETSA 112 PE R+E V C +C +GH F DGPKP + R+ I S+S++FV TS+ Sbjct: 22229 PERIRTE---VRCARCNAHMGHVF-EDGPKPTRKRYCINSASIEFVNADPATSS 22378 Score = 35.3 bits (108), Expect = 0.29 Identities = 18/45 (40%), Positives = 26/45 (57%) Frame = +1 Query: 68 KVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFVPKGKETSA 112 +V C +C +GH F +DGP P + RF I S+S+ FV + A Sbjct: 20458 EVRCSRCSAHMGHVF-DDGPPPKHRRFCINSASIDFVKSATPSKA 20589
>gnl|Dsim-washu-w501-asm|Contig20.125 Length = 16361 Score = 41.5 bits (130), Expect = 0.004 Identities = 18/44 (40%), Positives = 24/44 (54%) Frame = +3 Query: 12 FQNHFEPGVYVCAKCGYELFSSRSKYAHSSPWPAFTETIHADSV 55 + H+E GVY C C +LFSS +KY WPAF + + V Sbjct: 4344 YNKHYEKGVYQCIVCHQDLFSSDTKYDSGCGWPAFNDVLDKGKV 4475 Score = 37.6 bits (116), Expect = 0.062 Identities = 23/54 (42%), Positives = 32/54 (59%) Frame = +1 Query: 59 PEHNRSEALKVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFVPKGKETSA 112 PE R+E V C +C +GH F DGPKP + R+ I S+S++FV TS+ Sbjct: 7255 PERIRTE---VRCARCNAHMGHVF-EDGPKPTRKRYCINSASIEFVNADPATSS 7404 Score = 35.3 bits (108), Expect = 0.29 Identities = 18/45 (40%), Positives = 26/45 (57%) Frame = +2 Query: 68 KVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFVPKGKETSA 112 +V C +C +GH F +DGP P + RF I S+S+ FV + A Sbjct: 5453 EVRCSRCSAHMGHVF-DDGPPPKHRRFCINSASIDFVKSATPSKA 5584
>gi|27605048|emb|BX031767.1|CNS08WOB Single read from an extremity of a full-length cDNA clone made from Anopheles gambiae total adult females. 3-PRIME end of clone FK0AAA47BD04 of strain 6-9 of Anopheles gambiae (African malaria mosquito) Score = 75.5 bits (184), Expect = 3e-14 Identities = 38/100 (38%), Positives = 52/100 (52%), Gaps = 2/100 (2%) Frame = -2 Query: 12 FQNHFEPGVYVCAKCGYELFSSRSKYAHSSPWPAFTETIHADSVA--KRPEHNRSEALKV 69 + +E G Y+C C ELFSS +KY WPAF + + V K P +V Sbjct: 865 YNKFYEKGTYICVVCSQELFSSETKYDSGCGWPAFNDVLDQGKVTLHKDPSIPGRVRTEV 686 Query: 70 SCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFVPKGKE 109 C KC +GH F DGP P + R+ I S+S++F+P G E Sbjct: 685 RCSKCAAHMGHVF-EDGPPPTRKRYCINSASIEFMPAGSE 569
SECIS elements
gnl|Dsim-washu-w501-asm|Contig27.138: 1620 1527 (SECIS on complementary strand)
GAUAAGUGGU GGUAAUU AAUUAUUCAACUU AUGAA GAUUAUUUCUU AA AGGCCUCUGGCU CGGAAAUGGUC UGAA CUU UGUUGC ACACGCGAGG
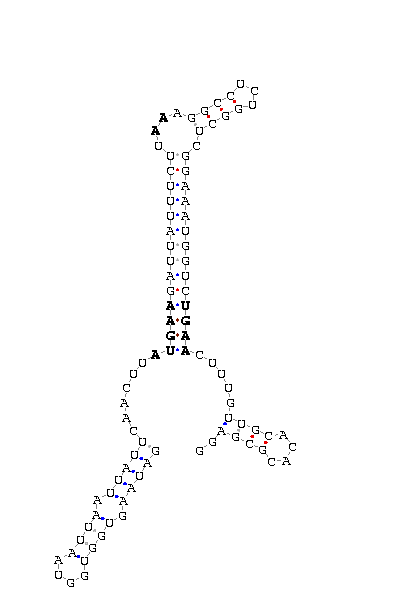
COVE score: 32.12 (Recommended threshold for COVE: 15)
gnl|Dyak-washu-assembly 040407|Contig8.19: 70796 70704 (SECIS on complementary strand)
GUUAAGUGAU ACAAAUU AAUUAUUCAACUU AUGAG GACUAUUUCUU AA AGGCCUUGGCU CGGAAAUAGUC UGAA CAU UAUUGU ACAUGAGAGG
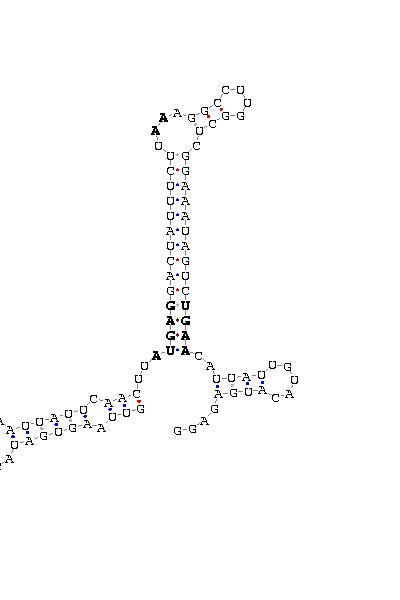
COVE score: 32.58 (Recommended threshold for COVE: 15)
Multiple alignment
If you want to see the ClustalW alignment, click here.

Unrooted N-J tree
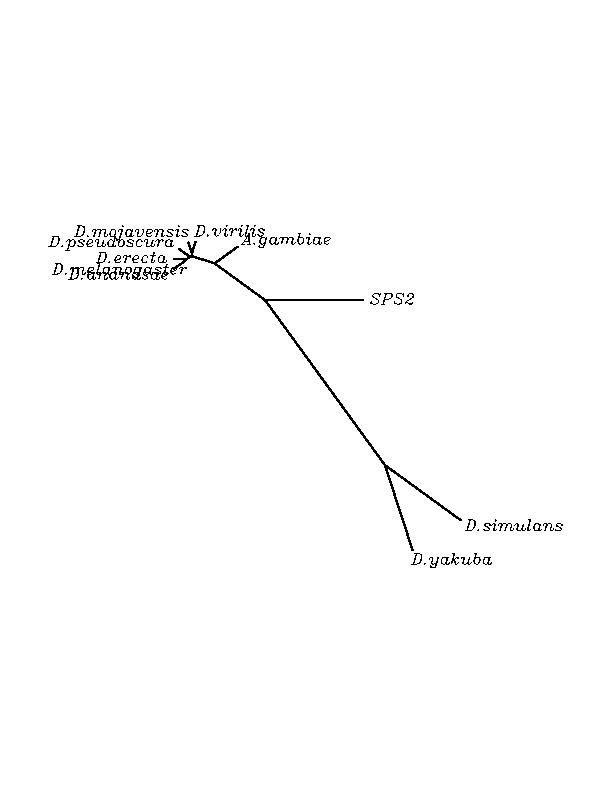
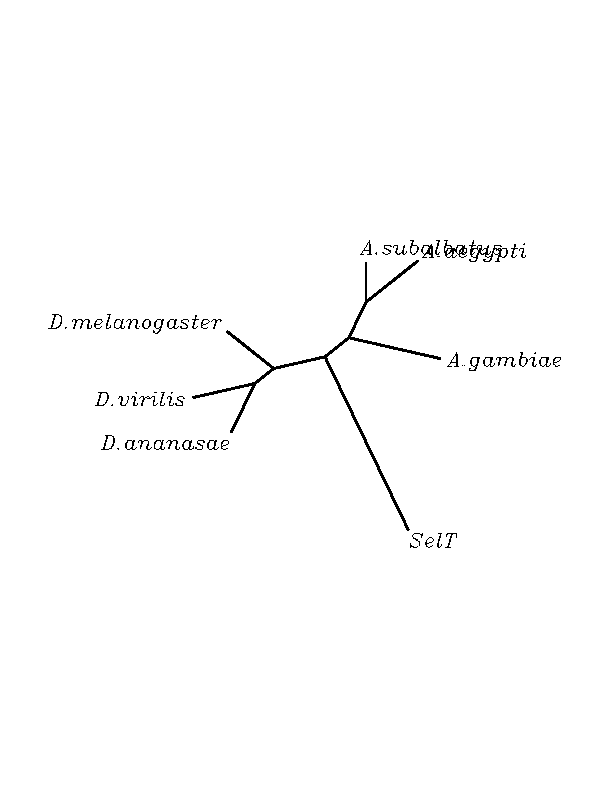
SelT
tblastn
>gnl|Dana-agencourt-040714-asm|contig_1097 3e-30 43% >gnl|Dvir-agencourt-run1029-asm|contig_992 5e-30 42% >4_group4 type=chromosome; loc=4_group4:1..6604331;ID=4_group4;release=r1.03; species=dpse 6e-30 40% >gi|24581818|ref|NM_135053.2| Drosophila melanogaster CG3887-PA (CG3887) mRNA, complete cds 2e-37 43% >gi|42765836|gb|AY440807.1| Armigeres subalbatus ASAP ID: 42399 putative: selenoprotein T mRNA. 1e-36 44% >gi|42761900|gb|AY433028.1| Aedes aegypti ASAP ID: 36405 putative: selenoprotein T mRNA. 7e-34 48% >gi|58393304|ref|XM_319979.2| Anopheles gambiae str.PEST ENSANGP00000016703 8e-33 45%
Gene prediction
If you want to check all the exonerate outputs in gff format, click here.
>gi|42765836|gb|AY440807.1| Armigeres subalbatus ASAP ID:42399 putative:selenoprotein T mRNA Score = 151 bits (381), Expect = 1e-36 Identities = 69/154 (44%), Positives = 101/154 (65%) Frame = +2 Query: 25 GPLLKFQICVS*GYRRVFEEYMRVISQRYPDIRIEGENYLPQPIYRHIASFLSVFKLVLI 84 G + F C S GYR+ F+EY +I ++YP+I I G NY P +++ L V KL++I Sbjct: 218 GATMTFMYCYSCGYRKAFDEYYNIIHEKYPEITIRGGNYDPPGFNMYLSKILLVTKLLMI 397 Query: 85 GLIIVGKDPFAFFGMQAPSIWQWGQENKVYACMMVFFLSNMIENQCMSTGAFEITLNDVP 144 ++ D F F PS W+W +NK+YACM+VFFL NM+E Q +S+GAFEI LNDVP Sbjct: 398 IALVSNFDLFGFLRQPMPSWWRWCTDNKIYACMLVFFLGNMLEAQLISSGAFEIALNDVP 577 Query: 145 VWSKLESGHLPSMQQLVQILDNEMKLNVHMDSIP 178 VW KLE+G +P+ Q+L QI+D+ ++ + ++ P Sbjct: 578 VWPKLETGRIPAPQELFQIIDSHLQFSDKIEQNP 679
>gi|42761900|gb|AY433028.1| Aedes aegypti ASAP ID: 36405 putative: selenoprotein T mRNA Score = 140 bits (353), Expect = 2e-33 Identities = 63/132 (47%), Positives = 90/132 (68%) Frame = +3 Query: 25 GPLLKFQICVS*GYRRVFEEYMRVISQRYPDIRIEGENYLPQPIYRHIASFLSVFKLVLI 84 G + F C S GYR+ ++EY +I ++YP+I I G NY P +++ V KL +I Sbjct: 324 GATMTFMYCYSCGYRKAYDEYYNIIHEKYPEITIRGANYDPPGFNMYLSKIXLVAKLAMI 503 Query: 85 GLIIVGKDPFAFFGMQAPSIWQWGQENKVYACMMVFFLSNMIENQCMSTGAFEITLNDVP 144 +++ + F F ++ PS WQW +NK+YACMMVFFL NM+E Q +S+GAFEI+LNDVP Sbjct: 504 MVLMSNFNLFGFLNLRIPSWWQWCTDNKMYACMMVFFLGNMLEAQLISSGAFEISLNDVP 683 Query: 145 VWSKLESGHLPS 156 VWSKLE+G +P+ Sbjct: 684 VWSKLETGRIPA 719
>gi|58393304|ref|XM_319979.2| Anopheles gambiae str. PEST ENSANGP00000016703, partial mRNA Score = 138 bits (348), Expect = 8e-33 Identities = 66/145 (45%), Positives = 93/145 (64%) Frame = +1 Query: 25 GPLLKFQICVS*GYRRVFEEYMRVISQRYPDIRIEGENYLPQPIYRHIASFLSVFKLVLI 84 G + F C S GYR+ F++Y +I ++YP+I I G NY P + ++ L V KL+LI Sbjct: 130 GATMTFLYCYSCGYRKAFDDYHNLILEKYPEITIRGSNYDPSGVNMLLSKVLLVTKLLLI 309 Query: 85 GLIIVGKDPFAFFGMQAPSIWQWGQENKVYACMMVFFLSNMIENQCMSTGAFEITLNDVP 144 ++ D + G WQW NK+YA MM+FFL N +E Q +S+GAFEITLNDVP Sbjct: 310 AALMSNYDIGRYIGNPFAGWWQWCFNNKLYASMMIFFLGNTLEAQLISSGAFEITLNDVP 489 Query: 145 VWSKLESGHLPSMQQLVQILDNEMK 169 VWSKLE+G P+ Q++ QI+DN ++ Sbjct: 490 VWSKLETGRFPAPQEMFQIIDNHLQ 564
SECIS elements
No SECIS elements were found by SeciSearch.
Multiple alignment
If you want to see the ClustalW alignment, click here.
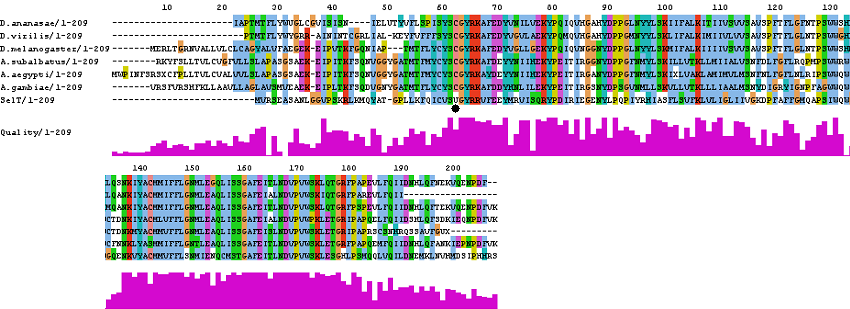
Unrooted N-J tree
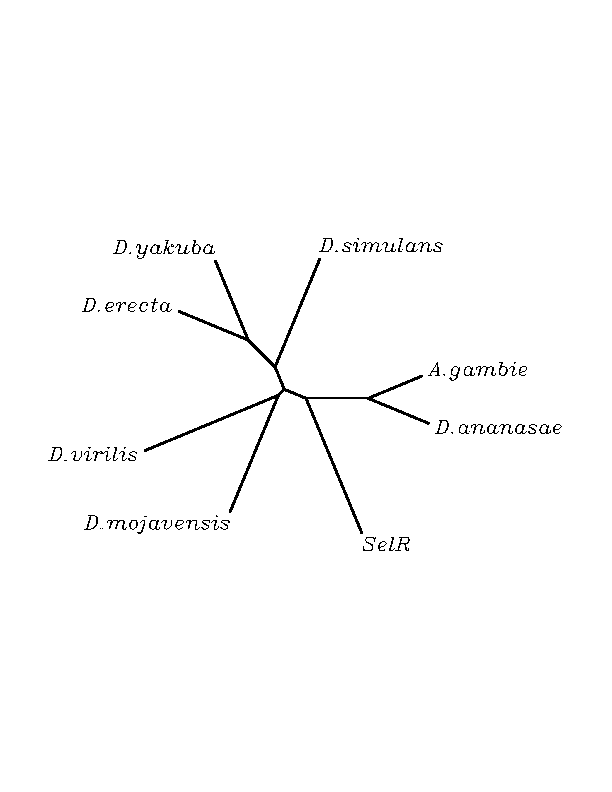
SelR
tblastn
>gnl|Dmov-agencourt-run0811-asm|contig_717 0.003 40% >gnl|Dana-agencourt-040714-asm|contig_4797 0.004 40% >gnl|Dyak-washu-assembly_040407|Contig7.13 0.004 40% >gnl|Dvir-agencourt-run1029-asm|contig_386 0.004 40% >2 type=chromosome; loc=2:1..30711475; ID=2; release=r1.03; species=dpse 0.004 40% >gnl|Dere-agencourt-run1028-asm|contig_1017 0.004 40% >gnl|Dsim-washu-w501-asm|Contig20.125 0.004 40% >gi|27605048|emb|BX031767.1|CNS08WOB Single read from an extremity of a full-length cDNA clone made from Anopheles gambiae total adult females.3-PRIME 3e-14 38%
Gene prediction
For the sequences that exonerate could not have been performed, tblastn outputs are shown:
>gnl|Dmov-agencourt-run0811-asm|contig_717 Length = 57670 Score = 41.8 bits (131), Expect = 0.003 Identities = 18/44 (40%), Positives = 24/44 (54%) Frame = -1 Query: 12 FQNHFEPGVYVCAKCGYELFSSRSKYAHSSPWPAFTETIHADSV 55 + H+E GVY C C +LFSS +KY WPAF + + V Sbjct: 21133 YNKHYEKGVYRCIVCHQDLFSSDTKYDSGCGWPAFNDVLDKGKV 21002 Score = 36.5 bits (112), Expect = 0.13 Identities = 21/46 (45%), Positives = 29/46 (63%) Frame = -3 Query: 59 PEHNRSEALKVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFV 104 PE R+E V C +C +GH F DGPKP + R+ I S+S++FV Sbjct: 18638 PERIRTE---VRCARCNAHMGHVF-EDGPKPTRKRYCINSASIEFV 18513 Score = 35.1 bits (107), Expect = 0.36 Identities = 18/37 (48%), Positives = 24/37 (64%) Frame = -2 Query: 68 KVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFV 104 +V C KC +GH F +DGP P + RF I S+S+ FV Sbjct: 20268 EVRCSKCSAHMGHVF-DDGPPPKHRRFCINSASIDFV 20161
>gnl|Dana-agencourt-040714-asm|contig_4797 Length = 61609 Score = 41.5 bits (130), Expect = 0.004 Identities = 18/44 (40%), Positives = 24/44 (54%) Frame = -3 Query: 12 FQNHFEPGVYVCAKCGYELFSSRSKYAHSSPWPAFTETIHADSV 55 + H+E GVY C C +LFSS +KY WPAF + + V Sbjct: 43103 YNKHYEKGVYQCIVCHQDLFSSDTKYDSGCGWPAFNDVLDKGKV 42972 Score = 40.4 bits (126), Expect = 0.009 Identities = 25/55 (45%), Positives = 34/55 (61%) Frame = -3 Query: 59 PEHNRSEALKVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFVPKGKETSAS 113 PE R+E V C +C +GH F DGPKP + R+ I S+S++FV K+ SAS Sbjct: 40394 PERIRTE---VRCARCSAHMGHVF-EDGPKPTRKRYCINSASIEFVTGEKDPSAS 40242 Score = 35.6 bits (109), Expect = 0.24 Identities = 27/70 (38%), Positives = 37/70 (52%), Gaps = 1/70 (1%) Frame = -3 Query: 36 KYAHSSPWPAFTETIHADSV-AKRPEHNRSEALKVSCGKCGNGLGHEFLNDGPKPGQSRF 94 K+ HSS TI + V +K R+E V C +C +GH F +DGP P + RF Sbjct: 42236 KFRHSSHTSITVNTIKSTVVISKTLGMVRTE---VRCSRCSAHMGHVF-DDGPPPKHRRF 42069 Query: 95 *IFSSSLKFV 104 I S+S+ FV Sbjct: 42068 CINSASIDFV 42039
>gnl|Dyak-washu-assembly_040407|Contig7.13 Length = 111046 Score = 41.5 bits (130), Expect = 0.004 Identities = 18/44 (40%), Positives = 24/44 (54%) Frame = +1 Query: 12 FQNHFEPGVYVCAKCGYELFSSRSKYAHSSPWPAFTETIHADSV 55 + H+E GVY C C +LFSS +KY WPAF + + V Sbjct: 46012 YNKHYEKGVYQCIVCHQDLFSSDTKYDSGCGWPAFNDVLDKGKV 46143 Score = 37.6 bits (116), Expect = 0.062 Identities = 23/54 (42%), Positives = 32/54 (59%) Frame = +1 Query: 59 PEHNRSEALKVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFVPKGKETSA 112 PE R+E V C +C +GH F DGPKP + R+ I S+S++FV TS+ Sbjct: 48979 PERIRTE---VRCARCNAHMGHVF-EDGPKPTRKRYCINSASIEFVNADPATSS 49128 Score = 35.3 bits (108), Expect = 0.29 Identities = 18/45 (40%), Positives = 26/45 (57%) Frame = +2 Query: 68 KVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFVPKGKETSA 112 +V C +C +GH F +DGP P + RF I S+S+ FV + A Sbjct: 47174 EVRCSRCSAHMGHVF-DDGPPPKHRRFCINSASIDFVKSATPSKA 47305
>gnl|Dvir-agencourt-run1029-asm|contig_386 Length = 191045 Score = 41.5 bits (130), Expect = 0.004 Identities = 18/44 (40%), Positives = 24/44 (54%) Frame = -1 Query: 12 FQNHFEPGVYVCAKCGYELFSSRSKYAHSSPWPAFTETIHADSV 55 + H+E GVY C C +LFSS +KY WPAF + + V Sbjct: 176732 YNKHYEKGVYQCIVCHQDLFSSDTKYDSGCGWPAFNDVLDKGKV 176601 Score = 36.5 bits (112), Expect = 0.13 Identities = 21/46 (45%), Positives = 29/46 (63%) Frame = -3 Query: 59 PEHNRSEALKVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFV 104 PE R+E V C +C +GH F DGPKP + R+ I S+S++FV Sbjct: 174108 PERIRTE---VRCARCNAHMGHVF-EDGPKPTRKRYCINSASIEFV 173983 Score = 35.6 bits (109), Expect = 0.24 Identities = 18/39 (46%), Positives = 25/39 (64%) Frame = -3 Query: 68 KVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFVPK 106 +V C KC +GH F +DGP P + RF I S+S+ FV + Sbjct: 175752 EVRCSKCSAHMGHVF-DDGPPPKHRRFCINSASIDFVKR 175639
>2 type=chromosome; loc=2:1..30711475; ID=2; release=r1.03; species=dpse Length = 30711475 Score = 41.5 bits (130), Expect = 0.004 BLAST HIT on Genome Map Identities = 18/44 (40%), Positives = 24/44 (54%) Frame = -3 Query: 12 FQNHFEPGVYVCAKCGYELFSSRSKYAHSSPWPAFTETIHADSV 55 + H+E GVY C C +LFSS +KY WPAF + + V Sbjct: 28012277 YNKHYEKGVYQCIVCHQDLFSSDTKYDSGCGWPAFNDVLDKGKV 28012146 Score = 36.5 bits (112), Expect = 0.13 BLAST HIT on Genome Map Identities = 21/46 (45%), Positives = 29/46 (63%) Frame = -1 Query: 59 PEHNRSEALKVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFV 104 PE R+E V C +C +GH F DGPKP + R+ I S+S++FV Sbjct: 28009705 PERIRTE---VRCARCNAHMGHVF-EDGPKPTRKRYCINSASIEFV 28009580 Score = 35.9 bits (110), Expect = 0.20 BLAST HIT on Genome Map Identities = 22/46 (47%), Positives = 29/46 (63%) Frame = -2 Query: 68 KVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFVPKGKETSAS 113 +V C KC +GH F +DGP P + RF I S+S+ FV K T+AS Sbjct: 28011345 EVRCSKCSAHMGHVF-DDGPPPKHHRFCINSASIDFV-KSAPTAAS 28011214
>gnl|Dere-agencourt-run1028-asm|contig_1017 Length = 31091 Score = 41.5 bits (130), Expect = 0.004 Identities = 18/44 (40%), Positives = 24/44 (54%) Frame = +3 Query: 12 FQNHFEPGVYVCAKCGYELFSSRSKYAHSSPWPAFTETIHADSV 55 + H+E GVY C C +LFSS +KY WPAF + + V Sbjct: 19356 YNKHYEKGVYQCIVCHQDLFSSDTKYDSGCGWPAFNDVLDKGKV 19487 Score = 37.6 bits (116), Expect = 0.062 Identities = 23/54 (42%), Positives = 32/54 (59%) Frame = +2 Query: 59 PEHNRSEALKVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFVPKGKETSA 112 PE R+E V C +C +GH F DGPKP + R+ I S+S++FV TS+ Sbjct: 22229 PERIRTE---VRCARCNAHMGHVF-EDGPKPTRKRYCINSASIEFVNADPATSS 22378 Score = 35.3 bits (108), Expect = 0.29 Identities = 18/45 (40%), Positives = 26/45 (57%) Frame = +1 Query: 68 KVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFVPKGKETSA 112 +V C +C +GH F +DGP P + RF I S+S+ FV + A Sbjct: 20458 EVRCSRCSAHMGHVF-DDGPPPKHRRFCINSASIDFVKSATPSKA 20589
>gnl|Dsim-washu-w501-asm|Contig20.125 Length = 16361 Score = 41.5 bits (130), Expect = 0.004 Identities = 18/44 (40%), Positives = 24/44 (54%) Frame = +3 Query: 12 FQNHFEPGVYVCAKCGYELFSSRSKYAHSSPWPAFTETIHADSV 55 + H+E GVY C C +LFSS +KY WPAF + + V Sbjct: 4344 YNKHYEKGVYQCIVCHQDLFSSDTKYDSGCGWPAFNDVLDKGKV 4475 Score = 37.6 bits (116), Expect = 0.062 Identities = 23/54 (42%), Positives = 32/54 (59%) Frame = +1 Query: 59 PEHNRSEALKVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFVPKGKETSA 112 PE R+E V C +C +GH F DGPKP + R+ I S+S++FV TS+ Sbjct: 7255 PERIRTE---VRCARCNAHMGHVF-EDGPKPTRKRYCINSASIEFVNADPATSS 7404 Score = 35.3 bits (108), Expect = 0.29 Identities = 18/45 (40%), Positives = 26/45 (57%) Frame = +2 Query: 68 KVSCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFVPKGKETSA 112 +V C +C +GH F +DGP P + RF I S+S+ FV + A Sbjct: 5453 EVRCSRCSAHMGHVF-DDGPPPKHRRFCINSASIDFVKSATPSKA 5584
>gi|27605048|emb|BX031767.1|CNS08WOB Single read from an extremity of a full-length cDNA clone made from Anopheles gambiae total adult females. 3-PRIME end of clone FK0AAA47BD04 of strain 6-9 of Anopheles gambiae (African malaria mosquito) Score = 75.5 bits (184), Expect = 3e-14 Identities = 38/100 (38%), Positives = 52/100 (52%), Gaps = 2/100 (2%) Frame = -2 Query: 12 FQNHFEPGVYVCAKCGYELFSSRSKYAHSSPWPAFTETIHADSVA--KRPEHNRSEALKV 69 + +E G Y+C C ELFSS +KY WPAF + + V K P +V Sbjct: 865 YNKFYEKGTYICVVCSQELFSSETKYDSGCGWPAFNDVLDQGKVTLHKDPSIPGRVRTEV 686 Query: 70 SCGKCGNGLGHEFLNDGPKPGQSRF*IFSSSLKFVPKGKE 109 C KC +GH F DGP P + R+ I S+S++F+P G E Sbjct: 685 RCSKCAAHMGHVF-EDGPPPTRKRYCINSASIEFMPAGSE 569
SECIS elements
No SECIS elements were found by SeciSearch.
Multiple alignment
If you want to see the ClustalW alignment, click here.
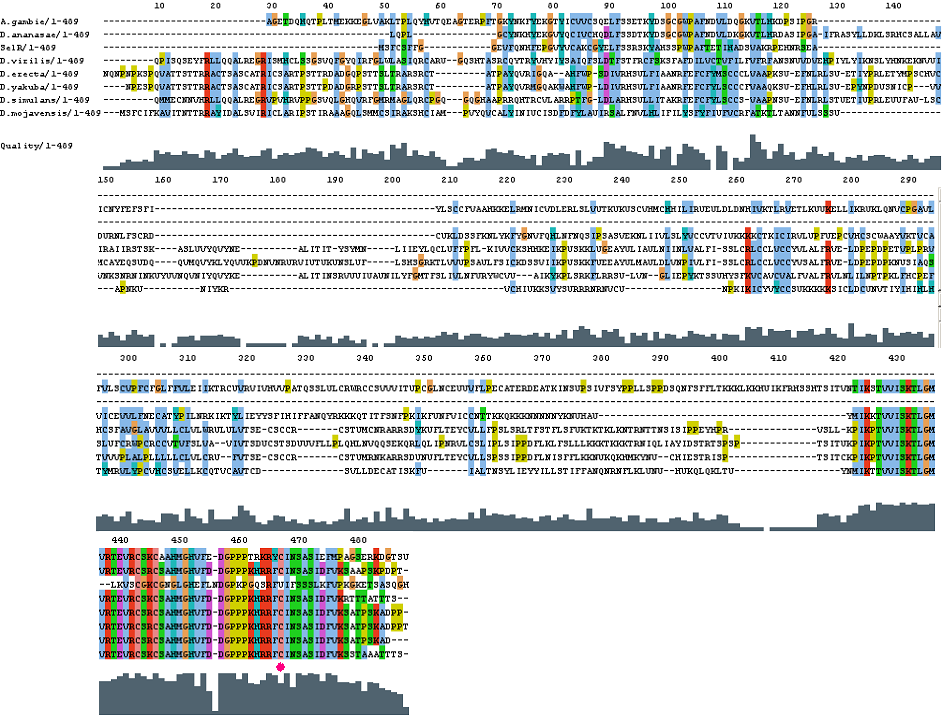
Unrooted N-J tree
GPx4
After tblastn similarity search,the all five human GPX selenoproteins, were found to match with the same DNA sequence in each of the species. This fact was probably due to the high similarity of the human GPx selenoproteins. Thus, we decided to determine, for each species, to which of the human selenoproteins was our common DNA sequence more closely related. To find out our best candidate we could have based on e-values and homology percentage shown in tblastn results, but in order to make our choice more confident we performed a multiple sequence aligment followed by an unrooted N-J tree, where we included all human GPx proteins and the protein we found for each of the species.
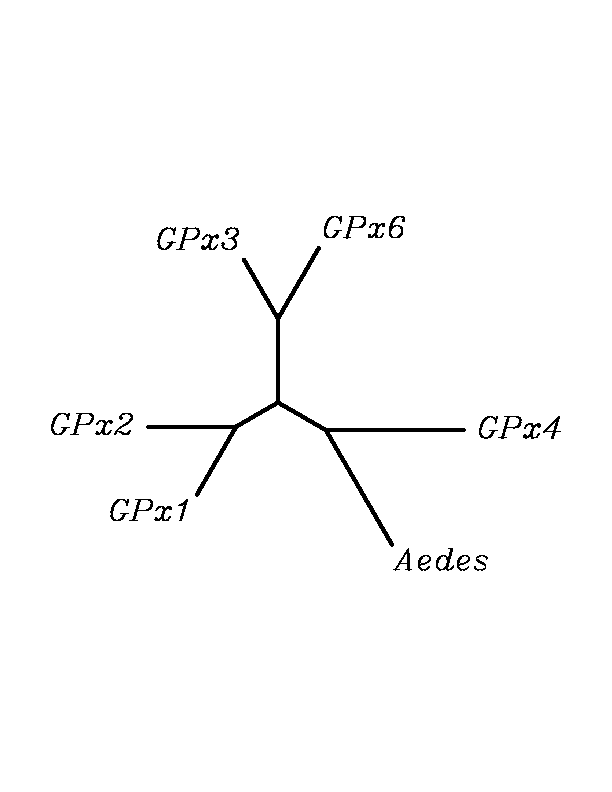
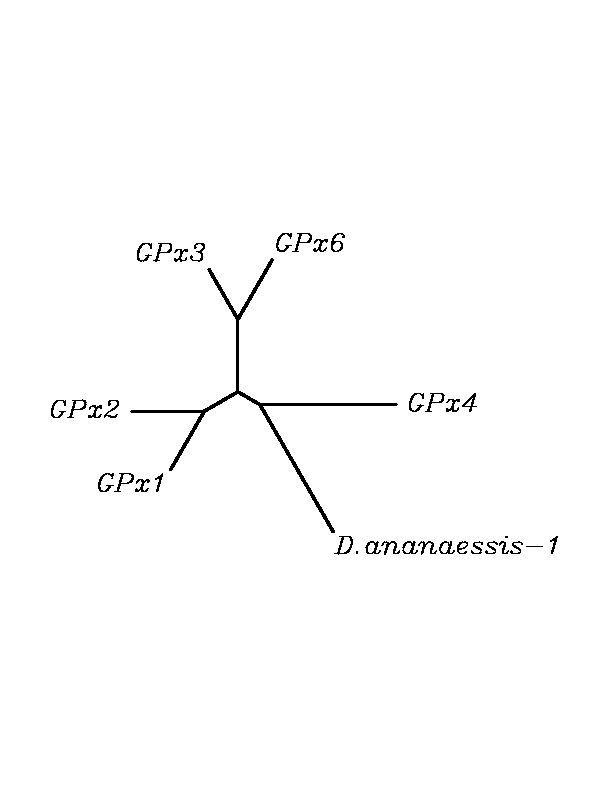
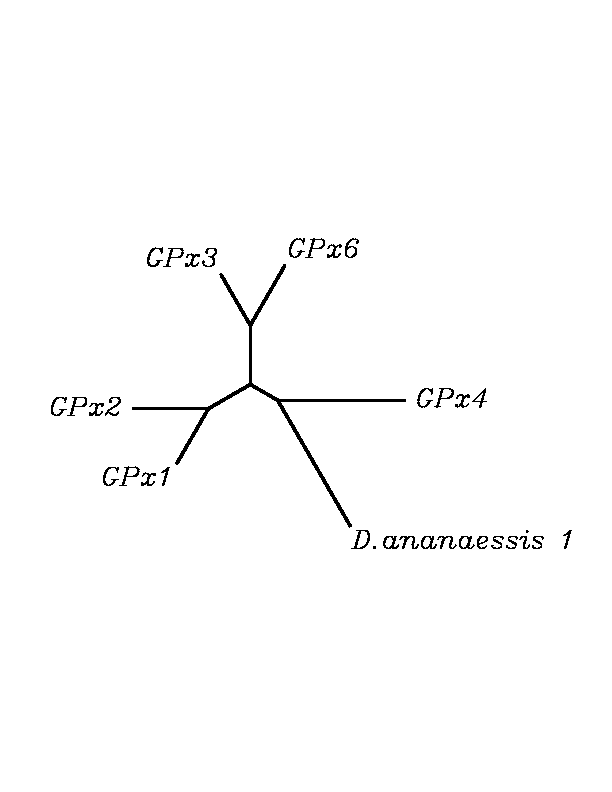
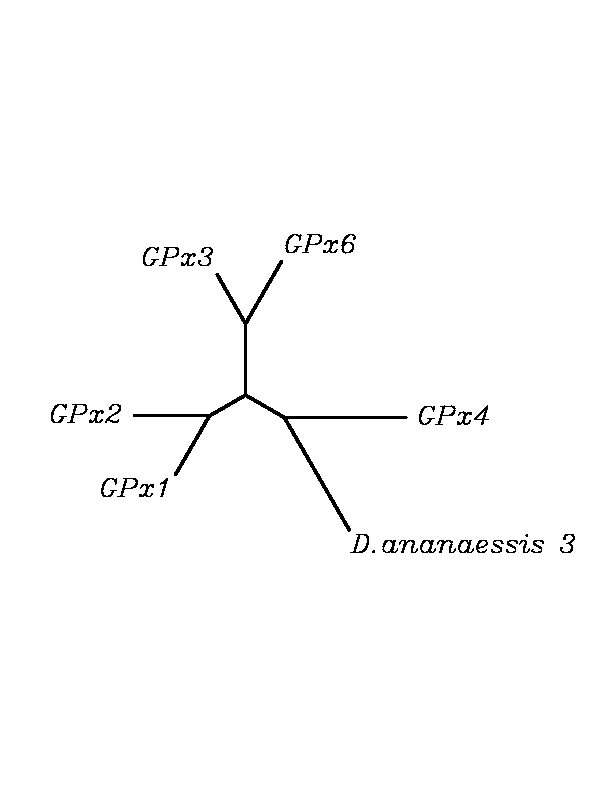
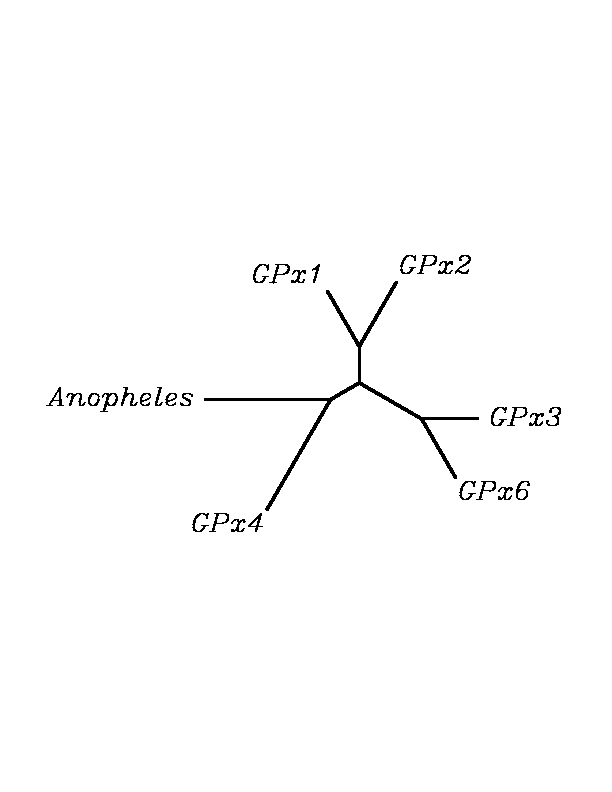
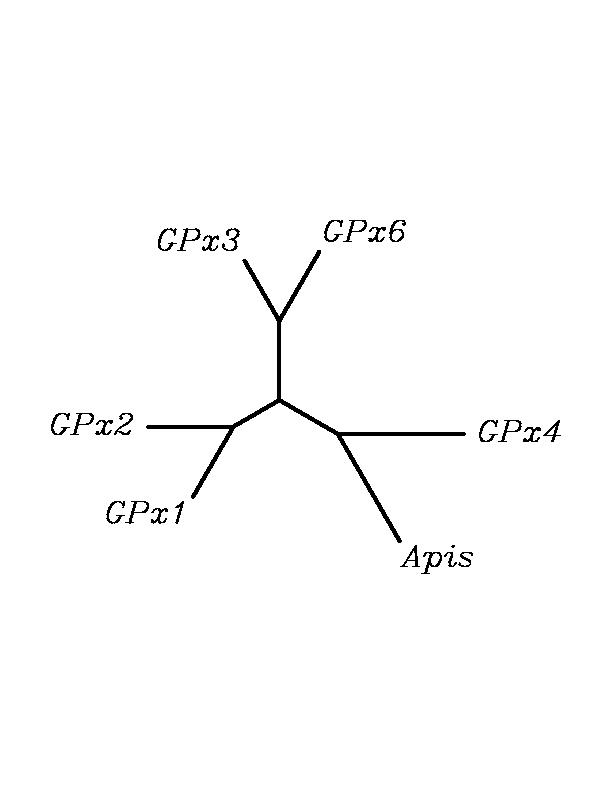
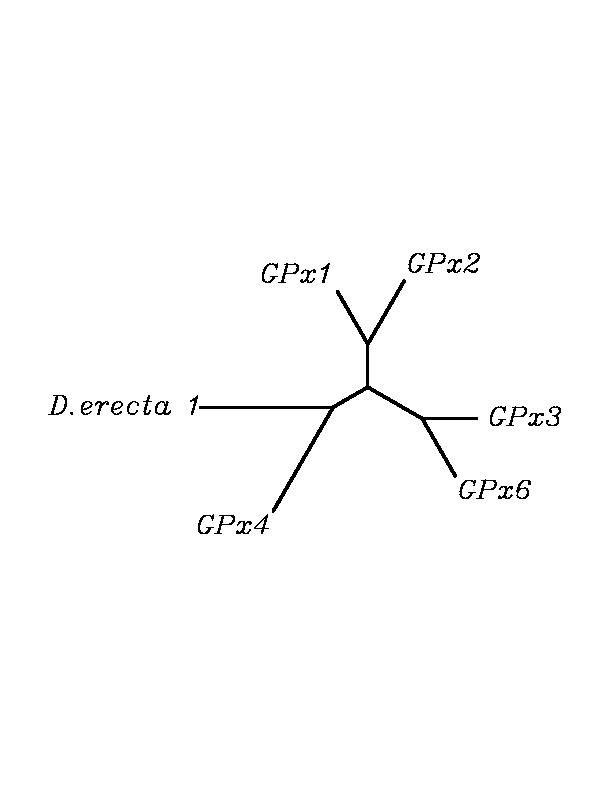
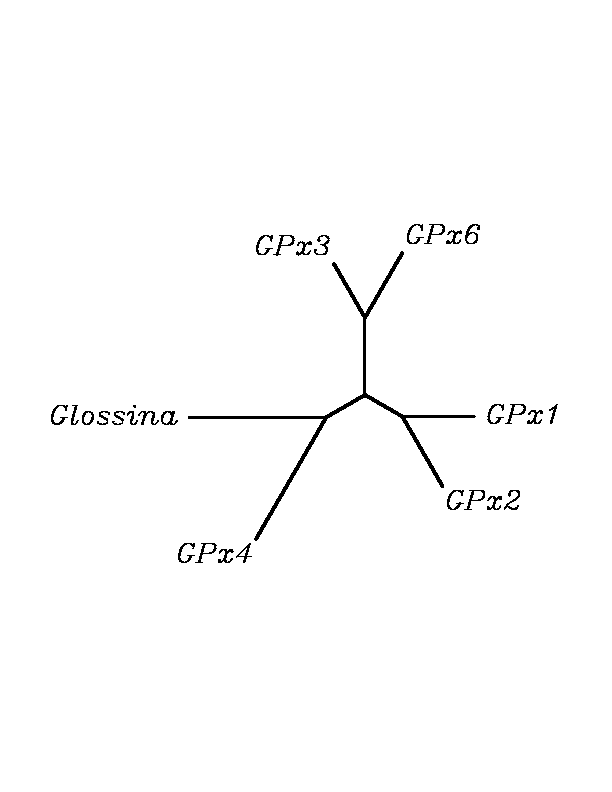
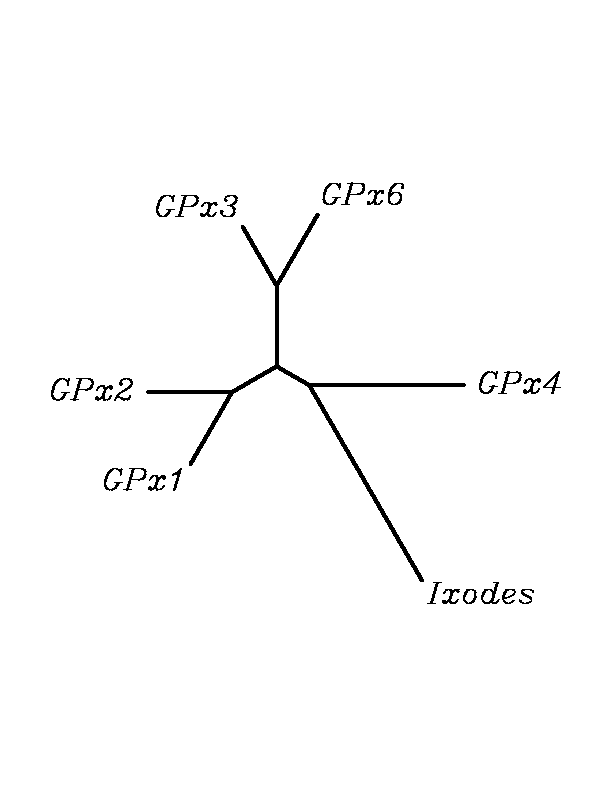
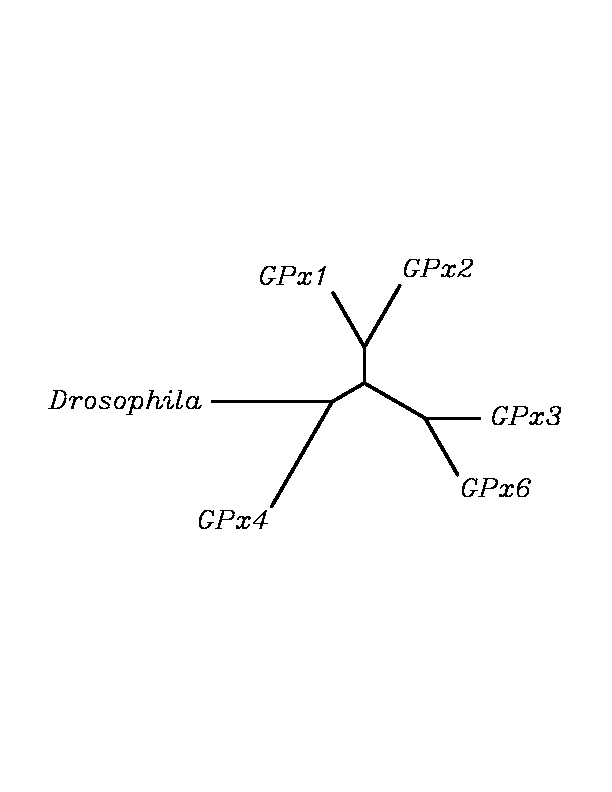
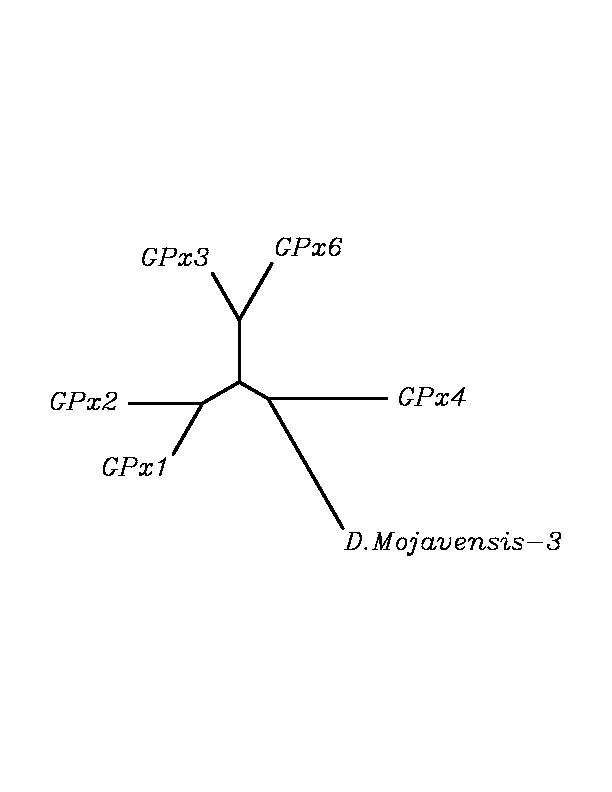
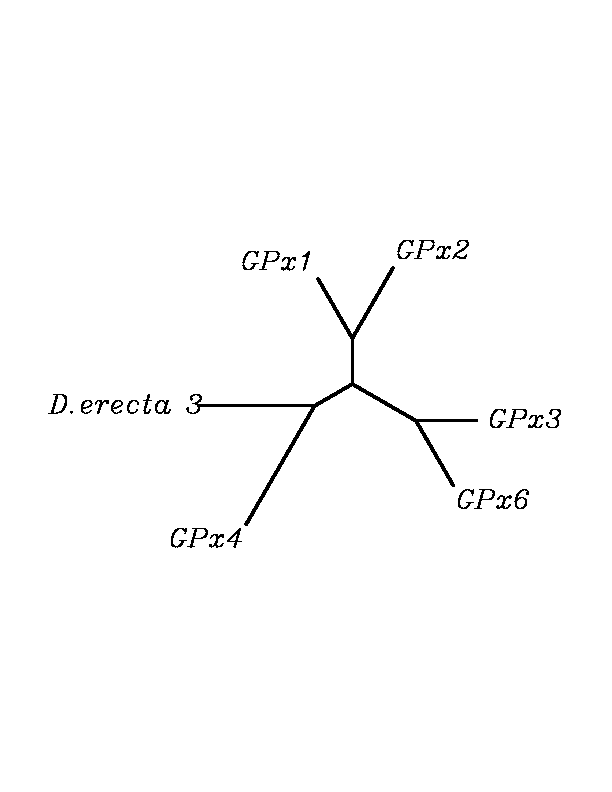
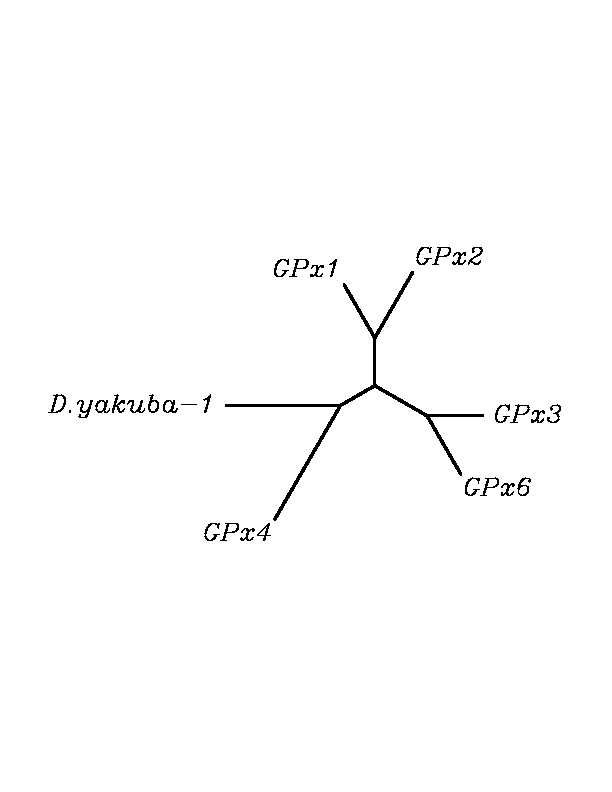
tblastn
As we can notice in the previous trees, all the proteins that we have studied are closer to GPx4 than to any other human GPx selenoprotein. Although this fact doesn't mean that all the proteins found in insect genomes correspond with human GPx4 function. Anyway, we will use this protein for gene prediction and multiple alingment and only GPx4 tblastn results will be shown.
>gnl|Dyak-washu-assembly_040407|Contig5.61 2e-25 46% >gnl|Dere-agencourt-run1028-asm|contig_2920 1e-24 39% >XR_group8 type=chromosome; loc=XR_group8:1..9190824 ID=XR_group release=r1.03; species=dpse 6e-23 38% >3 type=chromosome; loc=3:1..19738957;ID=3; release=r1.03; species=dps 1e-20 39% >gnl|Dsim-washu-w501-asm|Contig12.100 5e-14 39% >gnl|Dana-agencourt-040714-asm|contig_263 7e-14 49% >gnl|Dana-agencourt-040714-asm|contig_4443 3e-13 48% >gnl|Dana-agencourt-040714-asm|contig_1256 3e-13 39% >gnl|Dvir-agencourt-run1029-asm|contig_1983 1e-13 48% >gnl|Dere-agencourt-run1028-asm|contig_2847 1e-13 37% >gnl|Dsim-washu-w501-asm|Contig0.495 1e-13 46% >gnl|Dyak-washu-assembly_040407|Contig15.28 2e-12 38% >gnl|Dmov-agencourt-run0811-asm|contig_5053 3e-06 40% >gi|48117434|ref|XM_396418.1| Apis mellifera similar to putative thioredoxin perxidase 1e-47 55% >gi|33306812|gb|AF394234.1| Aedes aegypti glutathione peroxidase (GPx) mRNA, 2e-44 56% >gi|58384275|ref|XM_313166.2| Anopheles gambiae str. PEST ENSANGP00000024750 1e-34 46% >gi|50897528|gb|AY625510.1| Glossina morsitans morsitans clone Gmm0092 putative glutathione 6e-33 45% >gi|42765409|gb|AY440380.1| Armigeres subalbatus ASAP ID: 42545 glutathione peroxidase mRNA 5e-29 48% >gi|28564458|emb|AJ547804.1|IRI547804 Ixodes ricinus mRNA for glutathione peroxidase (gluper1) 1e-17 48% >gi|12958610|gb|AF321612.1| Venturia canescens virus-like particle protein mRNA 7e-18 33% >gi|40882422|gb|BT011331.1| Drosophila melanogaster SD18370 full insert cDNA 2e-34 44%
Gene prediction
If you want to check all the exonerate outputs in gff format, click here.
For the sequences that exonerate could not have been performed, tblastn outputs are shown:
>gnl|Dyak-washu-assembly_040407|Contig5.61(contig is not correct, exonerate can not be performed) Length = 146236 Score = 76.1 bits (253), Expect(2) = 2e-25 Identities = 40/86 (46%), Positives = 60/86 (69%) Frame = +1 Query: 30 ASRDDWRCARSMHEFSAKDIDGHMVNLDKYRGFVCIVTNVASQXGKTEVNYTQLVDLHAR 89 ++ D++ A S++EF+ KD G+ ++L+KY+G V +V N+AS+ G T+ NY +L DL + Sbjct: 63946 SANGDYKNAASIYEFTVKDTHGNDISLEKYKGKVVLVVNIASKCGLTKNNYQKLTDLKEK 64125 Query: 90 YAECGLRILAFPCNQFGKQEPGSNEE 115 Y E GL IL FPCNQFG Q P ++ E Sbjct: 64126 YGERGLVILNFPCNQFGSQMPEADGE 64203 Score = 60.1 bits (196), Expect(2) = 2e-25 Identities = 32/62 (51%), Positives = 44/62 (70%) Frame = +2 Query: 132 KICVNGDDAHPLWKWMKIQPKGKGILGNAIKWNFTKFLIDKNGCVVKRYGPMEEPLVIEK 191 ++ VNGDDA PL+K++K K G LG+ IKWNFTKFL++K G + RY P +P+ I K Sbjct: 64322 QVDVNGDDAAPLYKYLKA--KQTGTLGSGIKWNFTKFLVNKEGIPINRYAPTTDPMDIAK 64495 Query: 192 DL 193 D+ Sbjct: 64496 DI 64501
>gnl|Dere-agencourt-run1028-asm|contig_2920 Length = 10481 Score = 113 bits (385), Expect = 1e-24 Identities = 74/188 (39%), Positives = 110/188 (58%), Gaps = 24/188 (12%) Frame = +3 Query: 30 ASRDDWRCARSMHEFSAKDIDGHMVNLDKYRGFVCIVTNVASQXGKTEVNYTQLVDLHAR 89 ++ D++ A S++EF+ KD G+ ++L+KY+G V +V N+AS+ G T+ NY +L DL + Sbjct: 7311 SANGDYKNAASIYEFTVKDTHGNDISLEKYKGKVVLVVNIASKCGLTKNNYQKLTDLKEK 7490 Query: 90 YAECGLRILAFPCNQFGKQEPGSNEE----------------------IKEFAAGYNVKF 127 Y E GL IL FPCNQFG Q P ++ E +K +F Sbjct: 7491 YGERGLVILNFPCNQFGSQMPEADGEAMVCHLRDSKADIGEVFAKVRLLKLVVGCSRPRF 7670 Query: 128 D--MFSKICVNGDDAHPLWKWMKIQPKGKGILGNAIKWNFTKFLIDKNGCVVKRYGPMEE 185 + F ++ VNGD+A PL+K++K K G LG+ IKWNFTKFL++K G + RY P + Sbjct: 7671 NNLRFLQVDVNGDNAAPLYKYLK--AKQTGTLGSGIKWNFTKFLVNKEGVPINRYAPTTD 7844 Query: 186 PLVIEKDL 193 P+ I KD+ Sbjct: 7845 PMDISKDI 7868
>gnl|Dsim-washu-w501-asm|Contig12.100 Length = 22061 Score = 71.1 bits (235), Expect(2) = 5e-14 Identities = 48/122 (39%), Positives = 64/122 (52%), Gaps = 2/122 (1%) Frame = -2 Query: 74 GKTEVNYTQLVDLHARYAECGLRILAFPCNQFGKQEPGSN--EEIKEFAAGYNVKFDMFS 131 G T Y L L Y E GLRIL FPCNQFG Q P S+ E + + +F+ Sbjct: 8890 GLTLSQYNGLRYLLEEYEEQGLRILNFPCNQFGGQMPESDGQEMLDHLRREGANIGHLFA 8711 Query: 132 KICVNGDDAHPLWKWMKIQPKGKGILGNAIKWNFTKFLIDKNGCVVKRYGPMEEPLVIEK 191 KI V G A PL+K + + + I+WNF KFL+D+ G + KRYG EP+ + Sbjct: 8710 KIDVKGAQADPLYKLLTRHQ-------HDIEWNFVKFLVDRKGNIHKRYGAELEPVALTD 8552 Query: 192 DL 193 D+ Sbjct: 8551 DI 8546 Score = 26.9 bits (78), Expect(2) = 5e-14 Identities = 13/40 (32%), Positives = 24/40 (60%) Frame = -3 Query: 34 DWRCARSMHEFSAKDIDGHMVNLDKYRGFVCIVTNVASQX 73 D R ++H ++ +D G+ V LD + G V ++ N+AS+ Sbjct: 9072 DMRWRLTIHALTVRDTFGNPVQLDIFAGHVMLIVNIASR* 8953
>gnl|Dere-agencourt-run1028-asm|contig_2847 Length = 53404 Score = 70.2 bits (232), Expect(2) = 1e-13 Identities = 46/122 (37%), Positives = 64/122 (52%), Gaps = 2/122 (1%) Frame = +1 Query: 74 GKTEVNYTQLVDLHARYAECGLRILAFPCNQFGKQEPGSN--EEIKEFAAGYNVKFDMFS 131 G T Y L L Y + GLRIL FPCNQFG Q P S+ E + + +F+ Sbjct: 4213 GLTSSQYNGLRYLLEEYEDRGLRILNFPCNQFGGQMPESDGQEMLDHLRREGANIGHIFA 4392 Query: 132 KICVNGDDAHPLWKWMKIQPKGKGILGNAIKWNFTKFLIDKNGCVVKRYGPMEEPLVIEK 191 K+ V G A PL+K + + + I+WNF KFL+D+ G + KRYG EP+ + Sbjct: 4393 KVDVKGAQADPLYKLLTRHQQD-------IEWNFVKFLVDRKGNIHKRYGAELEPVALTD 4551 Query: 192 DL 193 D+ Sbjct: 4552 DI 4557 Score = 26.3 bits (76), Expect(2) = 1e-13 Identities = 13/42 (30%), Positives = 25/42 (59%) Frame = +3 Query: 34 DWRCARSMHEFSAKDIDGHMVNLDKYRGFVCIVTNVASQXGK 75 D R +++ ++ +D G V LDK+ G V ++ N+AS+ + Sbjct: 4038 DMRWRLTIQALTVRDTFGKPVQLDKFAGHVMLIVNIASK*ER 4163
>gnl|Dana-agencourt-040714-asm|contig_4443 Length = 7909 Score = 46.9 bits (149), Expect(2) = 3e-13 Identities = 27/56 (48%), Positives = 35/56 (62%) Frame = +1 Query: 60 RGFVCIVTNVASQXGKTEVNYTQLVDLHARYAECGLRILAFPCNQFGKQEPGSNEE 115 +G V +V N+ASQ G T+ NY L DL +Y + G IL FPCNQFG Q ++ E Sbjct: 1363 KGQVFLVVNIASQCGLTKNNYQTLTDLKEKYGDIG*IILNFPCNQFGSQMLETDGE 1530
>gnl|Dmov-agencourt-run0811-asm|contig_5053 Length = 2228 Score = 51.7 bits (166), Expect = 3e-06 Identities = 29/71 (40%), Positives = 46/71 (64%) Frame = -2 Query: 31 SRDDWRCARSMHEFSAKDIDGHMVNLDKYRGFVCIVTNVASQXGKTEVNYTQLVDLHARY 90 S D++ A S++EF+ KD G+ V+L+KY+G V ++ N+AS+ G T+ NY +L DL +Y Sbjct: 1636 SDGDYKNAASIYEFNVKDTHGNDVSLEKYKGQVILIVNIASKCGLTKNNYKKLTDLKEKY 1457 Query: 91 AECGLRILAFP 101 E G +P Sbjct: 1456 GERGTDHPELP 1424
>gnl|Dana-agencourt-040714-asm|contig_1256 Length = 119871 Score = 74.2 bits (246), Expect = 3e-13 Identities = 50/126 (39%), Positives = 67/126 (53%), Gaps = 2/126 (1%) Frame = -1 Query: 74 GKTEVNYTQLVDLHARYAECGLRILAFPCNQFGKQEPGSN-EEIKEFAAGYNVKF-DMFS 131 G T Y L +L +Y E GL IL FPCNQFG Q P S+ EI E +F+ Sbjct: 23997 GLTSSQYAGLHELREKYEERGLSILNFPCNQFGAQMPESDGREILEHLRQKKANIGHIFA 23818 Query: 132 KICVNGDDAHPLWKWMKIQPKGKGILGNAIKWNFTKFLIDKNGCVVKRYGPMEEPLVIEK 191 KI VNG +A PL+K + + I+WNF KFLID+ G + RYG ++P V+ Sbjct: 23817 KIKVNGRNADPLYKLLTRK-------APRIEWNFVKFLIDRKGNIYGRYGAEKKPAVLVN 23659 Query: 192 DLPHYF 197 D+ + Sbjct: 23658 DIERLL 23641
>gnl|Dyak-washu-assembly_040407|Contig15.28 Length = 56780 Score = 71.6 bits (237), Expect = 2e-12 Identities = 47/122 (38%), Positives = 64/122 (52%), Gaps = 2/122 (1%) Frame = -1 Query: 74 GKTEVNYTQLVDLHARYAECGLRILAFPCNQFGKQEPGSN--EEIKEFAAGYNVKFDMFS 131 G T Y L L Y + GLRIL FPCNQFG Q P S+ E + + +F+ Sbjct: 32579 GLTSTQYNGLRYLLEEYEDRGLRILNFPCNQFGAQMPESDGQEMLDHLRREGANIGQLFA 32400 Query: 132 KICVNGDDAHPLWKWMKIQPKGKGILGNAIKWNFTKFLIDKNGCVVKRYGPMEEPLVIEK 191 KI V G A PL+K + + + I+WNF KFL+D+ G + KRYG EP+ + Sbjct: 32399 KIDVKGAQADPLYKLLTRHQ-------HDIEWNFVKFLVDRRGNIYKRYGAELEPVALTD 32241 Query: 192 DL 193 D+ Sbjct: 32240 DI 32235
>gi|48117434|ref|XM_396418.1| Apis mellifera similar to putative thioredoxin perxidase (LOC412967), mRNA Score = 187 bits (476), Expect = 1e-47 Identities = 91/165 (55%), Positives = 120/165 (72%), Gaps = 1/165 (0%) Frame = +1 Query: 34 DWRCARSMHEFSAKDIDGHMVNLDKYRGFVCIVTNVASQXGKTEVNYTQLVDLHARYAEC 93 +W+ A ++++F AKDI G+ V+L+KYRG VCI+ NVAS G T+ NY +LV L+ +Y E Sbjct: 163 NWKSASTIYDFHAKDIHGNDVSLNKYRGHVCIIVNVASNCGLTDTNYRELVQLYEKYNEK 342 Query: 94 -GLRILAFPCNQFGKQEPGSNEEIKEFAAGYNVKFDMFSKICVNGDDAHPLWKWMKIQPK 152 GLRILAFP N+FG QEPG++ EI EF YNV FD+F KI VNGD+AHPLWKW+K Q Sbjct: 343 EGLRILAFPSNEFGGQEPGTSVEILEFVKKYNVTFDLFEKINVNGDNAHPLWKWLKTQ-- 516 Query: 153 GKGILGNAIKWNFTKFLIDKNGCVVKRYGPMEEPLVIEKDLPHYF 197 G + + IKWNF+KF+I+K G VV R+ P +PL +E +L YF Sbjct: 517 ANGFITDDIKWNFSKFIINKEGKVVSRFAPTVDPLQMESELKKYF 651
>gi|33306812|gb|AF394234.1| Aedes aegypti glutathione peroxidase (GPx) mRNA, complete cds Score = 177 bits (448), Expect = 2e-44 Identities = 91/160 (56%), Positives = 119/160 (74%), Gaps = 2/160 (1%) Frame = +1 Query: 40 SMHEFSAKDIDGHMVNLDKYRGFVCIVTNVASQXGKTEVNYTQLVDLHARYAEC-GLRIL 98 S+++FSA DIDG+ V+ ++YRG V I+ NVAS+ G T +Y +L +L+ Y E GLRIL Sbjct: 274 SVYDFSAVDIDGNKVDFERYRGHVLIIVNVASKCGYTAGHYKELNELYEEYGETEGLRIL 453 Query: 99 AFPCNQFGKQEPGSNEEIKEFA-AGYNVKFDMFSKICVNGDDAHPLWKWMKIQPKGKGIL 157 AFPCNQFG QEPG+NEEIK FA KFD+F+KI VNGD+AHPLW+++K Q +G G L Sbjct: 454 AFPCNQFGNQEPGTNEEIKHFARVEKGAKFDLFAKIYVNGDEAHPLWQFLK-QRQG-GTL 627 Query: 158 GNAIKWNFTKFLIDKNGCVVKRYGPMEEPLVIEKDLPHYF 197 +AIKWNFTKF++DKNG V+R+GP PL + +L YF Sbjct: 628 FDAIKWNFTKFIVDKNGQPVERHGPQTSPLQLRDNLKKYF 747
>gi|58384275|ref|XM_313166.2| Anopheles gambiae str. PEST ENSANGP00000024750 (ENSANGG00000011473), mRNA Score = 144 bits (364), Expect = 1e-34 Identities = 76/163 (46%), Positives = 109/163 (66%), Gaps = 2/163 (1%) Frame = +1 Query: 33 DDWRCARSMHEFSAKDIDGHMVNLDKYRGFVCIVTNVASQXGKTEVNYTQLVDLHARYAE 92 +D++ A+S+++F+ KD G V+L+KYRG V ++ N+ASQ G T+ NY +L +L +YA+ Sbjct: 118 EDYKNAKSVYDFTVKDSQGADVSLEKYRGKVLLIVNIASQCGLTKGNYAELTELSQKYAD 297 Query: 93 CGLRILAFPCNQFGKQEP-GSNEEIKEFAAGYNVKF-DMFSKICVNGDDAHPLWKWMKIQ 150 +IL+FPCNQFG Q P G EE+ + D+F+KI VNGD AHPL+K++K Sbjct: 298 KDFKILSFPCNQFGGQMPEGDGEEMVCHLRSAKAEVGDVFAKIDVNGDGAHPLYKYLK-- 471 Query: 151 PKGKGILGNAIKWNFTKFLIDKNGCVVKRYGPMEEPLVIEKDL 193 K G LG++IKWNF KFL++K+G V RY P P I KD+ Sbjct: 472 HKQGGTLGDSIKWNFAKFLVNKDGQPVDRYAPTTSPSSIVKDI 600
>gi|50897528|gb|AY625510.1 Glossina morsitans morsitans clone Gmm0092 putative glutathione peroxidase mRNA Score = 139 bits (350), Expect = 6e-33 Identities = 74/161 (45%), Positives = 107/161 (66%), Gaps = 5/161 (3%) Frame = +3 Query: 38 ARSMHEFSAKDIDGHMVNLDKYRGFVCIVTNVASQXGKTEVNYTQLVDLHARYAECGLRI 97 A S+++F+ KD G+ V+L++YRG V ++ N+ASQ G T+ NY +L DL +Y + GL+I Sbjct: 312 ASSIYDFTVKDTYGNDVSLEQYRGHVVLIVNIASQCGLTKNNYKKLTDLREKYGDKGLKI 491 Query: 98 LAFPCNQFGKQEPGSNEE-----IKEFAAGYNVKFDMFSKICVNGDDAHPLWKWMKIQPK 152 L FPCNQFG Q P S+ E +++ A D+F K+ VNG +A PL++++K K Sbjct: 492 LNFPCNQFGSQMPESDGEPMVCHLRDAKADIG---DVFQKVDVNGANAAPLYQYLK--AK 656 Query: 153 GKGILGNAIKWNFTKFLIDKNGCVVKRYGPMEEPLVIEKDL 193 G L +AIKWNFTKFL++K G VKRY P +P+ I KD+ Sbjct: 657 QGGTLVSAIKWNFTKFLVNKEGIPVKRYAPTTDPMDIAKDI 779
>gi|28564458|emb|AJ547804.1|IRI547804 Ixodes ricinus mRNA for glutathione peroxidase (gluper1 gene) Length = 914Score = 88.6 bits (218), Expect = 1e-17
Identities = 46/94 (48%), Positives = 65/94 (69%), Gaps = 1/94 (1%)
Frame = +1
Query: 27 TMCASRDDWRCARSMHEFSAKDIDGHMVNLDKYRGFVCIVTNVASQXGKTEVNYTQLVDL 88 T+ S + W+ A+S++EFSA DIDG+ V+ +KYRG V + NVA + T+ +Y +L L Sbjct: 298 TLANSDEGWKNAKSIYEFSALDIDGNKVDFNKYRGHVTQIVNVACKCLLTQEHYKKLSAL 477
Query: 87 HARYAEC-GLRILAFPCNQFGKQEPGSNEEIKEF 119 + +Y+E GLRI+AFP N F KQEP + EIKEF Sbjct: 478 YHKYSESKGLRIMAFPTNDFAKQEPWAEPEIKEF 579
Score = 107 bits (268), Expect = 2e-23
Identities = 54/97 (55%), Positives = 63/97 (64%)
Frame = +2
Query: 101 PCNQFGKQEPGSNEEIKEFAAGYNVKFDMFSKICVNGDDAHPLWKWMKIQPKGKGILGNA 160 P + PG N + F ++V FDMFSKI VNGD+AHPLWK++K K G L NA Sbjct: 524 PPTTLPSRNPGPNPRSRSFVKQFDVTFDMFSKISVNGDNAHPLWKYLK--EKQPGFLFNA 697
Query: 161 IKWNFTKFLIDKNGCVVKRYGPMEEPLVIEKDLPHYF 197 IKWNFTKFL+DKNG VKRYGP + IE DL YF Sbjct: 698 IKWNFTKFLVDKNGQPVKRYGPTDSFETIEADLLKYF 808
>gi|12958610|gb|AF321612.1|AF321612 Venturia canescens virus-like particle protein mRNA, complete cds Length = 861
Score = 89.4 bits (220), Expect = 7e-18
Identities = 57/168 (33%), Positives = 99/168 (58%), Gaps = 10/168 (5%)
Frame = +1
Query: 34 DWRCARSMHEFSAKDIDGHMVNLDKYRGFVCIVTNV---ASQXGKTEVNYTQLVDLHARY 90 DW+ A+S+++F+A +IDG ++NL+KY+G I+ N A+Q G +Y +L +L+ + Sbjct: 340 DWKTAKSLYQFTATNIDGDLINLNKYKGRPLIILNASSKANQLGTDMDHYEELKELYDKL 519
Query: 91 --AECGLRILAFPCNQF--GKQEPGSNEEIKEF-AAGYNVKFDMFSKICVNGDDAHPLWK 145 ++ L+ILAF CNQF ++ +N + KEF ++ D+F+K+ V G+ A PLWK Sbjct: 520 KGSKNELKILAFLCNQFDDSDKKDETNVDFKEFITTDKKLEADLFTKVEVTGEGAQPLWK 699
Query: 146 WMKIQPKGKGILGNA--IKWNFTKFLIDKNGCVVKRYGPMEEPLVIEK 19 W+ Q + + I +FT F++DK G + R+ ++ IE+ Sbjct: 700 WLYEQYCTDIDVTDCKEINHDFTIFVVDKMGHYLGRFEYTKDLSSIER 843
SECIS elements
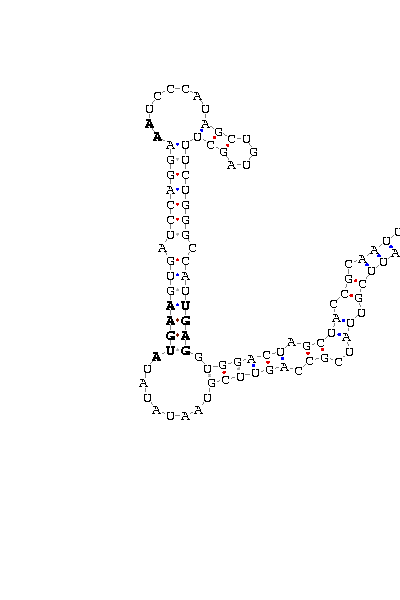
gnl|Dana-agencourt-040714-asm|contig 1256: 769 868
AAAUUCGUUA UCGCCAG UUCGUAAUAUAU AUGAA GUGAUCCAGGA AA UCCCAUAGCUGUAGCU UUCUGGGCCAU UGAG GUGGA CUAGCUA CCGCAAUUUA
COVE score: 13.44 (Recommended threshold for COVE: 15
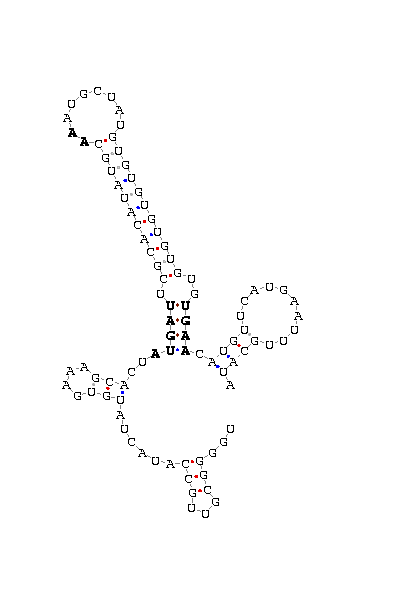
gnl|Dvir-agencourt-run1029-asm|contig 1983: 22898 22990
UGGGGCGUUG CCAUACU AUGUGAAAGCACU AUGAU UCGCACAUAUGC AA AUGCUAU GUGUGUGUGUGU GUGAA CAU GUUCAUG AAUUUGCAUA
COVE score: 6.42 (Recommended threshold for COVE: 15)
Multiple alignment
After checking GPx4 tblastn results we noticed some problematic cases in the following genomic fragments:
>gnl|Dana-agencourt-040714-asm|contig_1256(ana-1)
>gnl|Dyak-washu-assembly_040407|Contig15.28(yak-1)
>gnl|Dere-agencourt-run1028-asm|contig_2847(ere1)
>gnl|Dsim-washu-w501-asm|Contig12.100(sim-2)
As seen in the tblastn results related to the sequences above,although having good E-values, there is no evidence of matching the human"sec"( Stop codon).Even though, we realized that the flanking aminoacids were matching but in different frames.After performing exonerate analysis, no results were obtained.In order to overcome this problems, we proceded to find protein sequences using FASTACHUNK and TRANSLATE TOOL.
Regarding to the common aminoacid sequence found in the rest of genomic fragments, we concluded that our protein sequence was divided in two frames, so we adapt our candidate proteins with fragments coming from different frames and we submit them to multiple alignment analysis. Arguments explaining this fact will be given in discussion section.
If you want to see the ClustalW aligment, click here.
Unrooted N-J tree
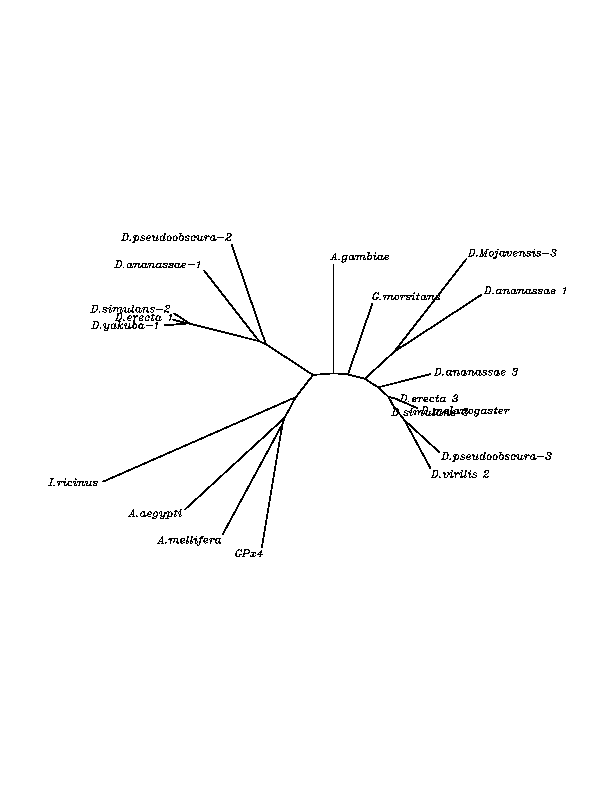
15 KDa
tblasn
>gnl|Dmov-agencourt-run0811-asm|contig_1424 2e-22 42% >gnl|Dvir-agencourt-run1029-asm|contig_3670 1e-21 39% >gnl|Dana-agencourt-040714-asm|contig_267 2e-21 51% >gnl|Dere-agencourt-run1028-asm|contig_376 2e-21 51% >gnl|Dyak-washu-assembly_040407|Contig17.16 6e-21 53% >gnl|Dsim-washu-w501-asm|Contig3419.1 6e-21 53% >XR_group6 type=chromosome; loc=XR_group6:1..6604477; ID=XR_group6;release=r1.03; species=dpse 6e-20 42% >gi|24666044|ref|NM_140743.1| Drosophila melanogaster CG7484-PB(CG7484)mRNA 5e-27 45% >gi|58386839|ref|XM_315090.2| Anopheles gambiae str.PEST ENSANGP00000011457 2e-32 44% >gi|18389880|gb|AF457547.1| Anopheles gambiae selenoprotein mRNA,partial cds 1e-31 44% >gi|42765457|gb|AY440428.1| Armigeres subalbatus ASAP ID:40327 selenoprotein mRNA 3e-31 43% >gi|42763702|gb|AY431560.1| Aedes aegypti ASAP ID:35705 selenoprotein mRNA 8e-30 45% >gi|48094317|ref|XM_394140.1| Apis mellifera similar to CG7484-PB(LOC410663),mRNA 3e-20 38%
Gene prediction
If you want to check all the exonerate outputs in gff format, click here.
For the sequences that exonerate could not have been performed, tblastn outputs are shown:
>gi|18389880|gb|AF457547.1| Anopheles gambiae selenoprotein mRNA, partial cds Score = 134 bits (337), Expect = 1e-31 Identities = 67/152 (44%), Positives = 95/152 (62%), Gaps = 4/152 (2%) Frame = +1 Query: 15 LRLLLATVL--QAVSAFGAEFSSEACRELGFXXXXXXXXX-XXXGQFNLLQLDPDCRGCC 71 +RL T L V+ GAEFS+E CRELG + L++L C CC Sbjct: 1 MRLFAITCLLFSIVTVIGAEFSAEDCRELGLIKSQLFCSACSSLSDYGLIELKEHCLECC 180 Query: 72 QEEAQFETK-KLYAGAILEVCGXKLGRFPQVQAFVRSDKPKLFRGLQIKYVRGSDPVLKL 130 Q++ + ++K K+Y A+LEVC K G +PQ+QAF++SD+P F L IKYVRG DP++KL Sbjct: 181 QKDTEADSKLKVYPAAVLEVCTCKFGAYPQIQAFIKSDRPAKFPNLTIKYVRGLDPIVKL 360 Query: 131 LDDNGNIAEELSILKWNTDSVEEFLSEKLERI 162 +D+ G + E LSI KWNTD+V+EF +L ++ Sbjct: 361 MDEQGTVKETLSINKWNTDTVQEFFETRLAKV 456
>gi|42765457|gb|AY440428.1| Armigeres subalbatus ASAP ID: 40327 selenoprotein mRNA Score = 132 bits (333), Expect = 3e-31 Identities = 68/157 (43%), Positives = 100/157 (63%), Gaps = 2/157 (1%) Frame = +3 Query: 8 CLVPAFGLRLLLATVLQAVSAFGAEFSSEACRELGFXXXXXXXXX-XXXGQFNLLQLDPD 66 C++ F L L+ V+ AEF+++ CRELGF G++ L +L Sbjct: 105 CIMNKFLLSLIPVLVI-VFQHTKAEFTTKDCRELGFIESQLFCSSCDTLGEYGLDELKDH 281 Query: 67 CRGCCQEEAQFETKKL-YAGAILEVCGXKLGRFPQVQAFVRSDKPKLFRGLQIKYVRGSD 125 CR CCQ++A+ K + Y A+LEVC K G +PQ+QAF++SD+P+ F L IKYVRG D Sbjct: 282 CRECCQKDAESSGKLMVYPKAVLEVCTCKFGVYPQIQAFIKSDRPQKFPNLTIKYVRGLD 461 Query: 126 PVLKLLDDNGNIAEELSILKWNTDSVEEFLSEKLERI 162 P++KL+D++GN+ E LSI KWNTD+V+EF +L ++ Sbjct: 462 PIVKLMDESGNVKETLSITKWNTDTVQEFFETRLTKV 572
>gi|42763702|gb|AY431560.1| Aedes aegypti ASAP ID: 35705 selenoprotein mRNA Score = 128 bits (321), Expect = 8e-30 Identities = 61/134 (45%), Positives = 89/134 (66%), Gaps = 2/134 (1%) Frame = +1 Query: 31 AEFSSEACRELGFXXXXXXXXX-XXXGQFNLLQLDPDCRGCCQEEAQFETKKL-YAGAIL 88 AEF+++ CR+LGF G++ L +L CR CCQ++ + K + Y A+L Sbjct: 196 AEFTAKDCRDLGFIKSQLYCSSCGTLGEYGLDELKDHCRECCQKDVESTGKLMVYPKAVL 375 Query: 89 EVCGXKLGRFPQVQAFVRSDKPKLFRGLQIKYVRGSDPVLKLLDDNGNIAEELSILKWNT 148 EVC K G +PQ+QAF++SD+P+ F L IKYVRG DP++KL+D+ GN+ E LSI KWNT Sbjct: 376 EVCTCKFGAYPQIQAFIKSDRPQKFPNLTIKYVRGLDPIVKLMDEAGNVKETLSITKWNT 555 Query: 149 DSVEEFLSEKLERI 162 D+V+EF +L ++ Sbjct: 556 DTVQEFFETRLTKV 597
>gi|48094317|ref|XM_394140.1| Apis mellifera similar to CG7484-PB (LOC410663), mRNA Score = 96.7 bits (239), Expect = 3e-20 Identities = 53/136 (38%), Positives = 77/136 (56%), Gaps = 2/136 (1%) Frame = +1 Query: 26 VSAFGAEFSSEACRELGFXXXXXXXXXXXXGQFNLLQLDPDCRGCCQEEAQFETK--KLY 83 V+ EFS++ C+ LGF + NLL C ++ K Y Sbjct: 22 VNIVSTEFSADDCKSLGF------------NKANLL---------CSTYDDYDASGLKRY 138 Query: 84 AGAILEVCGXKLGRFPQVQAFVRSDKPKLFRGLQIKYVRGSDPVLKLLDDNGNIAEELSI 143 A+LEVC K G +PQ+QAF++S++P ++ LQIKYVRG DP++KL D + + + L I Sbjct: 139 PRAVLEVCTCKFGAYPQIQAFIKSNRPNKYKNLQIKYVRGLDPIIKLFDADNKVEDILDI 318 Query: 144 LKWNTDSVEEFLSEKL 159 KW+TDSV+EFL+ L Sbjct: 319 HKWDTDSVDEFLATHL 366
SECIS elements
gnl|Dvir-agencourt-run1029-asm|contig 3670: 11575 11669
UAGUACGAAG GCAUGCG CGUCUGGGAGAGC AUGAU GGCUGACUUUA AA GACGGCAGUCGA
GAAGUCACGC UGAA AGGA UCACAGU GGAACAUAGA
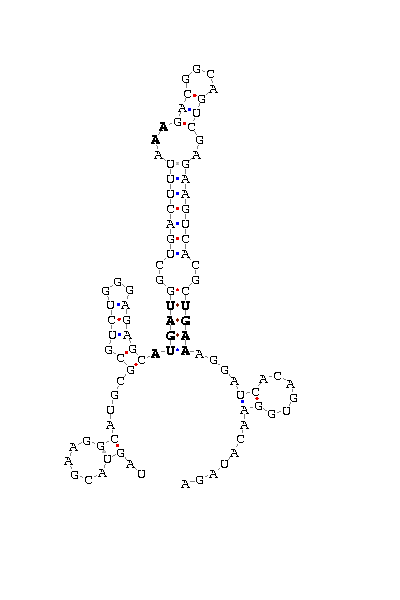
COVE score: 18.15 (Recommended threshold for COVE: 15)
Multiple alignment
If you want to see the clustalW alignment, click here.
Unrooted N-J tree
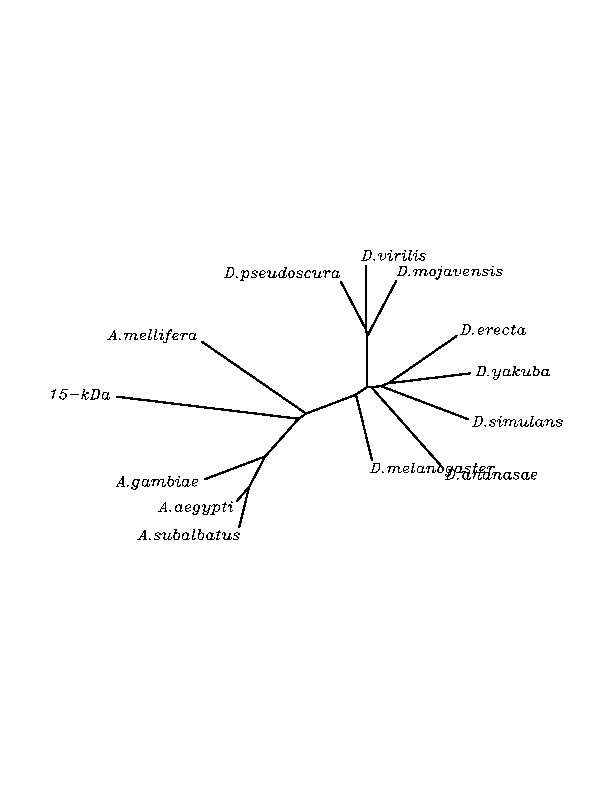
TR3
As in GPx family, after doing tblastn similarity search,all three TR proteins were found to match with the same DNA sequence in each of the species. Consequently, we decided to perform a multiple sequence aligment followed by an unrooted N-J tree as we did with GPx family.Inthis case we included all human TR proteins and the protein we found for each of the species.
tblastn
As we can notice in the previous figures all the proteins that we have studied are closer to TR3 than to any other human TR selenoprotein.Even though, this fact doesn't mean that all proteins found in insect genomes correspond with human TR3 function. Anyway, we will use this protein for gene prediction and multiple alingment, only TR3 tblastn results will be shown.
tblastn
>XR_group8 type=chromosome; loc=XR_group8:1..9190824; ID=XR_group8; release=r1.03; species=dpse e-149 54% >gnl|Dvir-agencourt-run1029-asm|contig_2338 e-147 53% >gnl|Dmov-agencourt-run0811-asm|contig_5619 e-146 53% >gnl|Dana-agencourt-040714-asm|contig_394 e-146 53% >gnl|Dyak-washu-assembly_040407|Contig131.3 e-145 53% >gnl|Dana-agencourt-040714-asm|contig_2492 e-142 53% >XL_group3a type=chromosome; loc=XL_group3a:1..2686958; ID=XL_group3a; release=r1.03; species=dpse e-140 50% >gnl|Dmov-agencourt-run0811-asm|contig_2707 e-139 53% >gnl|Dvir-agencourt-run1029-asm|contig_3163 e-138 53% >gnl|Dyak-washu-assembly_040407|Contig34.25 e-137 52% >gnl|Dere-agencourt-run1028-asm|contig_5904 e-101 52% >gnl|Dere-agencourt-run1028-asm|contig_278 1e-62 52% >gnl|Dsim-washu-w501-asm|Contig11.144 1e-29 46% >gnl|Dsim-washu-w501-asm|Contig2066.1 1e-19 50% >gi|33089107|gb|AY329357.1| Apis mellifera ligustica thioredoxin reductase (Trxr-1) mRNA, e-153 54% >gi|58380391|ref|XM_310514.2| Anopheles gambiae str. PEST ENSANGP00000017329 e-153 56% >gi|1848293|gb|U88187.1|MDU88187 Musca domestica glutathione reductase family member mRNA, e-150 53% >gi|27819972|gb|BT003266.1| Drosophila melanogaster LD21729 full insert cDNA e-147 53% >gi|42764077|gb|AY431890.1| Aedes aegypti ASAP ID: 42805 thioredoxin reductase mRNA sequence e-131 55% >gi|50897530|gb|AY625511.1| Glossina morsitans morsitans clone Gmm2366 putative thioredoxin e-101 50% >gnl|Dana-agencourt-040714-asm|contig_1097 3e-30 43% >gnl|Dvir-agencourt-run1029-asm|contig_992 5e-30 42% >4_group4 type=chromosome; loc=4_group4:1..6604331;ID=4_group4;release=r1.03; species=dpse 6e-30 40% >gi|24581818|ref|NM_135053.2| Drosophila melanogaster CG3887-PA (CG3887) mRNA, complete cds 2e-37 43% >gi|42765836|gb|AY440807.1| Armigeres subalbatus ASAP ID: 42399 putative: selenoprotein T mRNA. 1e-36 44% >gi|42761900|gb|AY433028.1| Aedes aegypti ASAP ID: 36405 putative: selenoprotein T mRNA. 7e-34 48% >gi|58393304|ref|XM_319979.2| Anopheles gambiae str.PEST ENSANGP00000016703 8e-33 45%
Gene prediction
For the sequences that exonerate could not have been performed, tblastn outputs are shown:
gnl|Dyak-washu-assembly_040407|Contig131.3(contig is not correct, exonerate can not be performed) Length = 18324 Score = 518 bits (1333), Expect = e-145 Identities = 261/486 (53%), Positives = 333/486 (68%), Gaps = 2/486 (0%) Frame = -1 Query: 38 DYDXXXXXXXXXXXACAKEAAQLGRKVAVVDYVEPSPQGTRWGLGGTCVNVGCIPKKLMH 97 DYD ACAKEAA G +V DYV+P+P GT+WG+GGTCVNVGCIPKKLMH Sbjct: 14415 DYDLVVLGGGSAGLACAKEAAGCGARVLCFDYVKPTPVGTKWGIGGTCVNVGCIPKKLMH 14236 Query: 98 QAALLGGLIQDAPNYGWEVA-QPVPHDWRKMAEAVQNHVKSLNWGHRVQLQDRKVKYFNI 156 QA+LLG + +A YGW V Q + DWRK+ +VQNH+KS+NW RV L+D+KV+Y N Sbjct: 14235 QASLLGEAVHEAVAYGWNVDDQNLRPDWRKLVRSVQNHIKSVNWVTRVDLRDKKVEYVNS 14056 Query: 157 KASFVDEHTVCGVAKGGKEIL-LSADHIIIATGGRPRYPTHIEGALEYGITSDDIFWLKE 215 SF D HT+ VA G E ++++++++A GGRPRYP I GA+E GITSDDIF + Sbjct: 14055 MCSFRDSHTIEYVAMPGAENRQVTSEYVVVAVGGRPRYPD-IPGAVELGITSDDIFSYER 13879 Query: 216 SPGKTLVVGASYVAWECAGFLTGIGLDTTIMMRTSPLRGFDQQMSSMVIEHMASHGTRFL 275 PG+TLVVGA YV ECA FL G+G + T+M+R+ LRGFD+QMS ++ M G FL Sbjct: 13878 EPGRTLVVGAGYVGLECACFLKGLGYEPTVMVRSIVLRGFDRQMSELLAAMMTERGIPFL 13699 Query: 276 RGCAPSRVRRLPDGQLQVTWEDSTTGKEDTGTFDTVLWAIGRVPDTRSLNLEKAGVDTSP 335 P V R DG+L V + ++TT K+ + FDTVLWAIGR LNLE AGV T Sbjct: 13698 GTTIPKAVERQADGRLLVRYHNTTTQKDGSDVFDTVLWAIGRKGLIEDLNLEAAGVKTHD 13519 Query: 336 DTQKILVDSREATSVPHIYAIGDVVEGRPELTPTAIMAGRLLVQRLFGGSSDLMDYDNVP 395 D KI+VD EATSVPHI+A+GD++ GRPELTP AI++GRLL +RLF GS+ LMDY +V Sbjct: 13518 D--KIVVDGAEATSVPHIFAVGDIIYGRPELTPVAILSGRLLARRLFAGSTQLMDYADVA 13345 Query: 396 TTVFTPLEYGCVGLSEEEAVARHGQEHVEVYHAHYKPLEFTVAGRDASQCYVKMVCLREP 455 TTVFTPLEY CVG+SEE A+ G +++EV+H +YKP EF + + CY+K V Sbjct: 13344 TTVFTPLEYSCVGMSEETAIELRGADNIEVFHGYYKPTEFFIPQKSVRHCYLKAVAEVSG 13165 Query: 456 PQLVLGLHFLGPNAGEVTQGFALGIKCGASYAQVMRTVGIHPTCSEEVVKLRISKRSGLD 515 Q +LGLH++GP AGEV QGFA +K G + ++ TVGIHPT +EE +L I+KRSG D Sbjct: 13164 DQKILGLHYIGPVAGEVIQGFAAALKSGLTVKTLLNTVGIHPTTAEEFTRLSITKRSGRD 12985 Query: 516 PTVTGC 521 PT C Sbjct: 12984 PTPASC 12967
SECIS elements
gnl|Dvir-agencourt-run1029-asm|contig 2338: 297811 297912
ACCCUGCAGC UUGAAAA GUUGUCUGUCCAU AUGAU AAGACAGGUAGA AA GAAAGCUGGCGAAAC UUCGAUUGUCUU UGAA AACAC AUUUGCA CAAGGCUAUU
COVE score: 9.87 (Recommended threshold for COVE: 15)
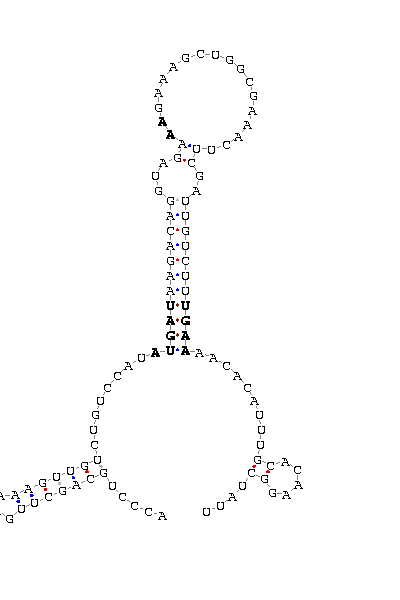
Multiple alignment
If you want to see the ClustalW alignment, click here.

Unrooted N-J tree
4. Discussion
The computational analysis as an attempt to decipher genomic contents emerges as a powerful tool, permiting new genes to be found. New programmes for gene prediction, similarity search systems and other tools are being developped and continously improved in a way that we can combine the ones that best fit to solve our specific problem. Our project was aimed at determining the presence or absence of selenoproteins in different insect genomes by using the various computational analysis resources that are nowadays available.
After having performed an exhaustive analysis by the methods mentioned above, we report evidence of candidate selenoproteins existing in insect genomes.
The results show presence of human selenoprotein orthologues in some insect species (on one hand D.ananassae, D.erecta, D.virilis and D.yakuba for selenoproteinH on the other hand D.simulans and D.yakuba for seleprotein SPS2).In addition to the mentioned results, we found many "Cys" orthologues and also many other proteins that even not having "Sec" (stop codon) or "Cys" their conservation level is extremely high.
THE SPS2 FAMILY
The overall conservation level of the obtained candidates was very high. We found that the majority of our candidates had an "Arg" residue matching with the human "Sec"(Stop codon) but two of the candidates had a Stop codon (candidate "Sec") matching to the human one. The later studies emphasized our hypothesis after finding that the Stop was in-frame according to the exonerate analysis and in addition, the Secis structures were present in the RNA sequence. We concluded that the proteins with "Arg" are other proteins belonging to the wide SPS family, and the other two with Stop codon are selenoproteins. The neighboor joinning tree associates these two proteins more closely to the human SPS2 selenoprotein than to the others even if they come from phylogenetically closer organisms.
THE H SELENOPROTEIN
Some candidate selenoproteins were found among the hits. 6 organisms had a stop codon matching with the human "Sec" and SECIS STRUCTURES with significant score were found in all of them. The stop codons were not in-frame according to the exonerate analysis, but we think that this could be due to the phylogenetic distance between the query human protein and the submitted genomes. It is likely that using the human selenoprotein as the template for the exonic structure moddeling of the insect genomic sequences not the most accurate approximation towards the real model.Moreover, in the multiple alignment, the Stop codon flanking aminoacids remain conserved among the species making us think that they are part of the protein what means part of the exonic structure.
THE R SELENOPROTEINS
No selenoprotein candidates were found among the hits. All the organisms were Cys homologous to the human selenoprotein and the region flanking this position seems to be the most conserved. Even though, with exception to this region conservation among sequences is barely found. This strongly suggests the presence of a common domain among all the submitted sequences. This is the probable reason why exonerate could not have been performed. The sequences just share a motif rather than being orthologous proteins. Nevertheless, if we focus in lower level empairing each sequence with the other in all the possible combinations, clusters of different similarity arise. Thus, the possibility of having orthology among some of them can not be discarded. No SECIS structures were found.
THE T SELENOPROTEINS
Orthologues of the human T selenoprotein were found in a wide variety of insect genomes. The multiple alignment showed that there are very conserved regions, and that the most of the protein's positions are identycal for all the sequences submitted. Eventhough, all the candidates had a Cys in the position that a "Sec" is found in the human selenoprotein, making us think that the the "Sec" was adquired in a later step of the evolution. The fact that the exonic model coming from the exonerate analysis does not include the Cys matching to the human "Sec" is probably due to the query used but not a real exclusion of this position from the real exonic region. No Secis structures were found.
THE GPX FAMILY
In this family 21 cys-homologous proteins in 15 different species were reported, all of them, according to the phylogenetic analysis, related to the human Gpx4.
Results coming from tblastn were quite significant, and some of them were further confirmed in the exonerate analysis were the human "Sec"matching Cys residues were placed in-frame. The multiple alignment showed a high conservation level, even in those sequences that could not have been submitted to exonerate.
As it was mentioned in the results section, although four of the genomic fragments had no reliable results after doing tblastn and exonerate, we reconstructed the proteins from two different frame sequences and submitted them to multiple aligment analysis. This revealed that our proteins are very well conserved among them and also towards the other sequences, what is more significant. This fact suggests that we are in front of four genes needing a frameshift to encode cys-homologous proteins. Thus, we speculate that these genes could have suffered an alternative 3' splice site. Even though, we could be facing a simple sequentiation mistake.
Our results also revealed another significant fact. Most of Drosophila species showed more than one cys-homologous protein:
D.ananassae : 3 cys-homologous
D.yakuba : 2 cys-homologous (one could not be confirmed due to a incorrect contig)
D.erecta: 2 cys-homologous
D.pseudoobscura: 2 cys-homologous
D.simulans: 2 cys-homologous
D.virilis: 1 cys-homologous
D.mojavensis: 1 cys-homologous
D.melanogaster: 1cys-homologous
Although all of these proteins are more closed to Gpx4 than to any other Gpx human protein, we can speculate that different cys-homologous proteins found for each species performe different functions knowing that at least, they are encoded by different genomic sequences.
Multiple sequence aligment followed by N-J unrooted tree suggested that the different cys-homologous proteins found in most of Drosophila species come from a duplication process. This is proved by the fact that in those species having more than one homologous protein, each of them is located in a different tree cluster. D.ananassae is supossed to have suffered two duplication process, while species showing just one cys-homologous protein are supossed not to have suffered any, and they are clustered together.
Independently of our results we have to bear in mind that not all the genomes that we have worked with are completely sequenced but D.melanogaster and D.pseudoobscura, so these results can not be considered as definitives.
A duplication event could drive to two different functional proteins as it has been mentioned above, but not necessarily. As it is known a duplication could yield a pseudogene, so unless performing more exhaustive and even experimental analysis, it will be impossible to confirm whether our cys-homologous candidates are functional or not.
Only two SECIS structures were found by SeciSearch and due to obtained scores, only the one corresponding to D.ananassae can be considered significant. Presence of SECIS in cys-homologous proteins represents an unusual case that is more deeply discussed at the end of this section..
THE 15KDA SELENOPROTEIN
We found 15Kda selenoprotein orthologues in almost every insect having a sequenced genome. The hits went up to 11 organisms in the similarity search, and it was obeserved that all of them had a Cys aminoacid at the position of the human "Sec". This fact made us think that non of them could be selenoproteins, but we found a significant Secis structure at the sequence belonging to D.virilisis. The score(18.5) is good enough to to think that a real Secis could be formed by this sequence, but the existence of a SECIS structure with no target codon to place a possible "Sec" would represent a not usual case. This could be explained from a model were an ancient selenoprotein with both SECIS and Sec substituted its Sec for a Cys but it has not passed time enough to make the SECIS dissapear.
THE TR FAMILY
In this family 20 cys-homologous proteins in 13 different species were reported, all of them, according to the phylogenetic analysis, related to the human TR3.
Results coming from tblastn were really significant, and most of them were further confirmed in the exonerate analysis were the human "Sec" matching Cys residues were placed in-frame. The multiple alignment showed a high conservation level, even in those sequences that could not have been submitted to exonerate.
As it has been observed with the GPX family, Drosophila species showed more than one cys-homologous candidate proteins. In this case all species but D.melanogaster showed 2 cys-homologous proteins.
Multiple sequence aligment followed by N-J unrooted tree suggested that the different cys-homologous proteins found in most of Drosophila species come from a duplication process. This is proved by the fact that in those species having more than one homologous protein, each ofthem is located in a different tree cluster.
As it was commented previuosly, we have to bear in mind that not all the genomes that we have worked with are completely sequenced but D.melanogaster and D.pseudoobscura, so these results can not be considered as definitives.
Although, as we have already commented, a duplication event could yield to a pseudogene expression, in this case, due to the high conservation among all sequences and the equal number of cys-homologous candidate proteins found, for most of the species, we can suggest, more consistently, that for Gpx family that we could be in front of two differents proteins. Anyway, unless performing more exhaustive and even experimental analysis, it will be impossible to confirm whether our cys-homologous candidates are functional or not.
Only one SECIS structure was found. Its score was not really significant.
The candidate selenoproteins found in this project represent probably a small sample of all the ones widespread in different organisms, so further genomic analysis need to be performed in order to establish their real importance. Appart from the obvious conclusions mentioned above, the unusual cases found make some evolutionary questions arise. The most likely is that proteins having Cys aminoacids suffered mutations that enabled their downstream 3'UTR region to form a SECIS structure.
Once having a SECIS structure a substitution of Cys by Sec would be affordable but not before, because that would lead to loose the translation of a larger or smaller part of the mRNA probably producing an unfunctional protein. Obviously, a change leading to an unfunctional protein would not be permitted by the natural selection.Nevertheless, formation of a SECIS element without any Sec to target seems difficult too. It is true that the UTR region is much less restrictive to mutations than the coding ones.
This makes this region more given to changes, including those needed to form a SECIS element, but it is difficult to believe that this kind of neutral changes would be accumulated and conserved until that point were the Cys would change into Sec and the SECIS would turn useful. Thus,two opposite logics make the evolutionary question of selenoprotein emergence look like the tale of the hen and the egg represented by the STOP codon (Sec) and the SECIS element.
5. References
ARTICLES:
- Castellano S, Morozova N, Morey M, Berry M, Serras F, Corominas M, Guigó R. In silico identification of novel
selenoproteins in the Drosophila melanogaster genome. EMBO reports 2, 697-702 (2001).
- Hatfield D, Gladyshev V. How selenium has altered our understanding of the genetic code.
Molecular and Cellular Biology 22. 3565-3576 (2002)
- S. Castellano, S.V. Novoselov, G.V. Kryukov, A. Lescure, E. Blanco, A. Krol. V.N. Gladyshev and R. Guigó.
Reconsidering the evolution of eukaryotic selenoproteins: a novel nonmammalian family with scattered phylogenetic distribution.
EMBO reports, 5(1):71-77 (2004)
- G.V. Kryukov, S. Castellano, S.V. Novoselov, A.V. Lobanov, O. Zehtab, R. Guigó and V.N. Gladyshev
Characterization of mammalian selenoproteomes.
Science, 300(5624):1439-1443 (2003)
6. Acknowledgements
We thank greatfully all our lecturers for making the world of Bioinformatics more accessible to us, specially Charles Chapple for supervising our job and having the patience of bearing us. We greatly thank the opportunity of discovering this world, a world that we will be pleased not to visit again to the end of our days. Thank you for making us work so hard, to the point that our girlfriends and boyfriends broke up for feeling abandoned. Thank you for making us smoke again, a habit that we managed to give up but we went back to, during this project. We greatly thank the greasy sandwiches from "la cafeteria del hospìtal" for feeding us with high vitaminic content food, essential for the highly demanding brain work that we had to perform. Finally, we thank our families for their unconditional support.