MÈTODES
Let's seXXXqs!!! és un programa desenvolupat en llenguatge Perl que ha estat dissenyat per a realitzar l'alineament global de dues seqüències d'aminoàcids o nucleòtids mitjançant l'algorisme de programació dinàmica de S.B. Needleman and C.D. Wunsch.
El programa requereix diversos paràmetres per realitzar l'alineament:
Un cop li hem introduït tota aquesta informació i l'executem, ens mostra per pantalla l'alineament òptim en format CLUSTAL.
*(Per veure el funcionament del programa de forma més detallada, podeu visitar l'apartat Disseny.
Quan volem alinear dues seqüències de símbols podem esperar una correspondència exacta, en la que una d'elles es troba replicada exactament dins l'altra, o bé una correspondència inexacta, on calcularem en quin grau les dues són similars. En Biologia veurem que les correspondències exactes molts cops no existeixen, de manera que haurem de trobar alguna manera per avaluar quina de les múltiples correspondències inexactes possibles entre dues seqüències és la millor, o sigui, quin és l'alineament òptim.
Per tal d'establir un criteri, s'assigna una puntuació o score a cada un dels alineaments possibles, i es determina que l'òptim és el que té la major puntuació. Aquesta puntuació resulta de la suma de puntuacions parcials de cada parella de símbols alineats; així, donarem una bona puntuació als símbols que s'assemblen i una dolenta als que no.
En l'àmbit de la biologia puntuarem favorablement aquells nucleòtids o aminoàcids que hagin variat poc durant l'evolució. És cert que no coneixem el procés evolutiu real, però assumirem que el més probable serà aquell que comporti menys canvis, o aquells més probables. Així, alinearem abans una Adenosina amb una altra Adenosina, que no pas amb una Citosina; però què escollirem a l'hora d'alinear una Adenosina amb una Citosina o amb una Guanina? A l'hora d'avaluar el grau de similutud entre símbols que no són iguals, ens regirem per la implicació del canvi de símbol: si ha suposat una transversió o una transició, si un aminoàcid polar s'ha canviat per un d'hidrofòbic... o senzillament per la probabilitat d'aquest canvi observada al llarg de l'evolució. Així s'han elaborat unes taules que assignen una puntuació específica per a cada aparellament de nucleòtids i aminoàcids respectivament: les matrius de substitució.
Tot i tenir un criteri per escollir quin d'entre tots és l'alineament òptim, se'ns presenta un problema logístic: donada l'habitual llargada de les seqüències de gens o proteïnes, la quantitat de possibilitats sol ser enormement gran, siguent poc pràctic i a vegades quasi impossible, haver de calcular-les totes. A més, hi ha un altre factor que fa augmentar el nombre de possibilitats, i és que també s'ha de considerar la possibilitat de que algun dels símbols no s'alineï, de forma que inserim un salt a l'altra seqüència. Aquest fenòmen és molt present en biologia mitjançant processos de deleció o inserció de nucleòtids al DNA, processaments alternatius del mRNA (splicing), modificacions post-traduccionals de proteïnes...
Gràcies a l'algorisme que S.B. Needleman i C.D. Wunsch idearen l'any 1970 aconseguim solventar aquest problema, doncs els càlculs es redueixen a 3 x v x w, siguent v la llargada d'una seqüència i w la de l'altra. Això s'aconsegueix descartant molts dels possibles alineaments, ja que l'alineament òptim es calcula de forma dinàmica a partir d'alineaments parcials, de la següent manera:
![]() Let's seXXXqs!!!
Let's seXXXqs!!!
PROGRAMACIÓ DINÀMICA
Així, per trobar l'alineament òptim entre dues seqüències de 100 símbols, hauríem de calcular aproximadament 10 elevat a 200 possibles alineaments!!!
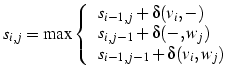
Es construeix una matriu S en la que s'enfronten les dues seqüències a alinear, esdevenint les mateixes seqüències les dimensions de la matriu; llavors, per a cada casella i,j de la matriu es calcula el valor de l'alineament parcial en aquell punt, el qual serà el màxim entre les tres opcions que ens presenta l'algorisme:
Entenem com "alineament parcial" l'alineament òptim aconseguit entre les subseqüències corresponents a cada una de les tres possibles caselles anteriors a (Si,j).
*(Per entendre millor el funcionament d'aquest procés, podeu visitar l'apartat Exemples de la plana web)
FORMAT FASTA
El format FASTA s'usa típicament per enregistrar seqüències i es caracteritza per mostrar una primera línia amb el símbol ">" precedint el nom identificador de la seqüència. A partir de la segona línia ja comença la seqüència en sí, dividida en 50, 60 o 70 símbols per línia. Els fitxers amb aquest format solen tenir l'extensió *.fa. A continuació es mostra un exemple:

MATRIUS DE SUBSTITUCIÓ
Les matrius de substitució més àmpliament emprades per a l'alineament de seqüències són aquelles que es fixen en els canvis observats durant l'evolució. En aquest àmbit trobem les matrius PAM i les BLOSUM com les més típiques:
Les matrius PAM s'acostumen a utilitzar per a seqüències que poden ser properes o que comparteixen un ancestre comú, doncs estan fetes a partir de proteïnes molt conservades. Per a seqüències més divergents, emprarem les PAM més elevades (80, 120, 250, 320...) doncs contemplaran períodes evolutius majors.
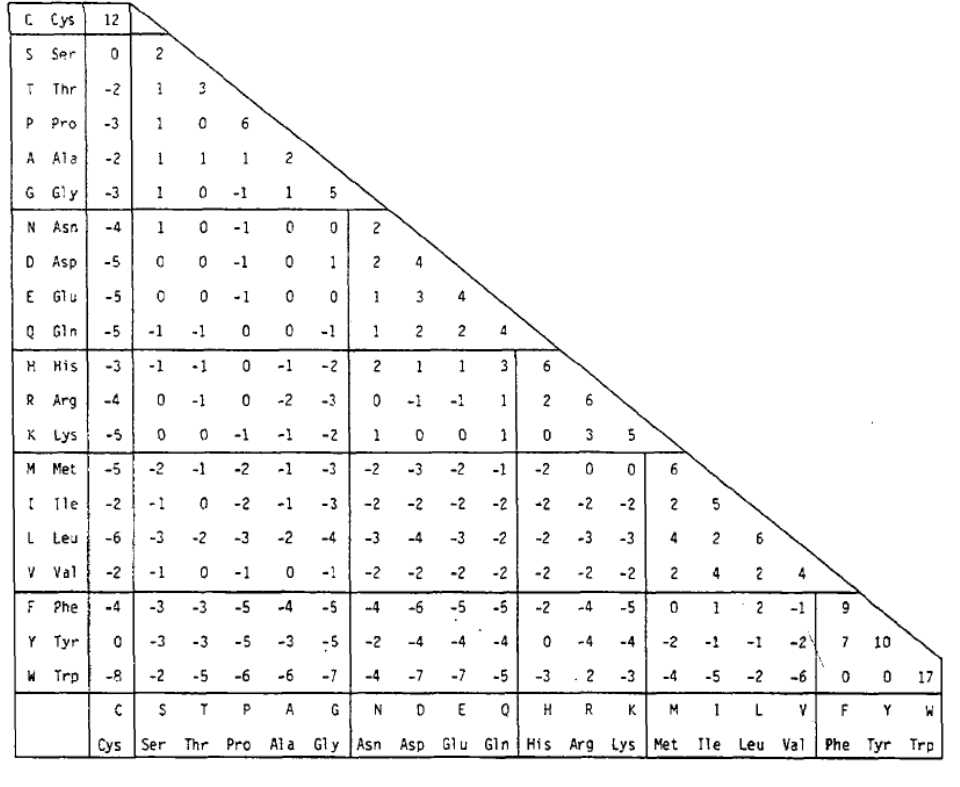
*Matriu PAM250. Al tractar-se del logaritme d'una raó de versemblança, els valors positius indicaran canvis favorables, i els negatius, desfavorables.
FORMAT CLUSTAL
El format CLUSTAL és un dels formats de sortida més emprats pels programes d'alineament. Mostra una capçalera amb el nom del format, la puntuació de l'alineament i els percentages d'identitat i similitud entre les dues seqüències alineades. A continuació ja es presenta l'alineament, amb la primera seqüència alineada sobre l'altra. Sota, hi trobem una línia amb un seguit de símbols, un per a cada aparellament: un asterisc (*) si els dos símbols alineats són iguals (identitat), dos punts (:) si el valor de substitució entre ells és superior a 0 (similitud), i un espai en blanc ( ) en tots els altres casos. Les seqüències alineades solen estar partides en línies de 50 o 60 símbols, que van precedides pel nom identificador (o pels seus 30 primers caracters, si és més llarg) més un espai. A continuació es mostra un exemple:
