{kind=link}
Per a poder validar els gens predits pels 3 programes usats, es fa còrrer les regions on s'han predit gens contra una base de dades d'EST humans (base de dades de seqüències codificants), utilitzant el programa BLAST , concretament MEGABLAST.
Així es podrà veure en quina mesura els gens predits s'assemblen a les seqüències de cDNA de la base de dades.
Es farà seguint els passos següents:
- Per tal de fer les prediccions més senzilles,es talla la seqüència en regions diferents, cadascuna de les quals engloba un o més gens.
- D'aquesta manera, a l'hora de comparar els gens predits amb els ESTs de la base de dades serà molt més fàcil, ja que s'està eliminant un cert soroll de fons.
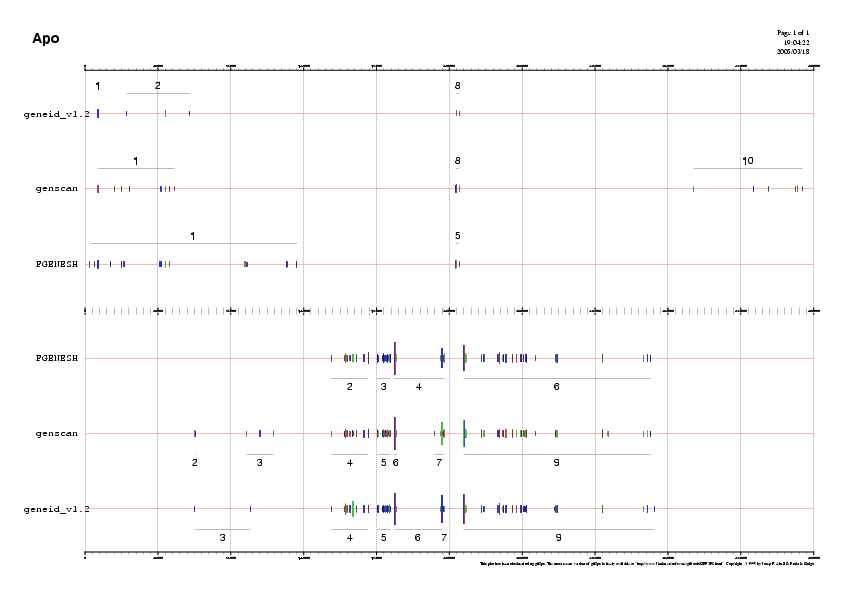
- Les regions s'han escollit segons la distribució dels gens predits, que es pot observar en la imatge on es troben totes les prediccions alhora ( Prediccio2.png ).
- Mirant la correspondència exònica existent entre els 3 programes, s'ha decidit considerar 7 regions diferents dins de la seqüència anònima.
- Les regions per a dur a terme l'anà comparatiu amb els ESTs es van escollir en funció de la distribució dels gens. És a dir,en una regió concreta vam observar quin era el gen que començava abans i vam tallar 1000nt upstream. El mateix vam fer amb el gen més llarg, a partir del qual vam tallar 1000nt downstream.
Amb això ens assegurem que la regió escollida tingui uns certs marges per evitar aliniaments inespecifics.
- Un cop decidides les regions, i determinats els seus marges, es va procedir a realitzar els talls usant la següent comanda:
<--"; ( echo ">"$REG"."$INI"-"$LEN ; fastachunk Apo.seq.masked $INI $LEN | fold -60 ) > Apo.valdacio.$REG.fa; }; done
- Aquesta comanda retorna l'inici i la longitut de cada regió que ha tallat i, a més la transforma a format fasta.
- Per dur a terme aquesta comanda s'ha de partir d'un fitxer tbl anomenat Seqüències.tbl que dóna la informació sobre : Regió/Inici/Final.
- Tenint ja totes les regions tallades, i amb les diferents subseqüències emmascarades i en format FASTA, es fa córrer un MEGABLAST marcant les opcións: EST_human, homo sapiens i pairwise.
- Això servirà per veure quin programa de prediccions ha donat les prediccions gèniques més acurades.
- Els resultats del blastn es troben en arxius txt i html:
Apo.Reg1.blast.est / Apo.Reg1.blast.html
Apo.Reg2.blast.est / Apo.Reg2.blast.html
Apo.Reg3.blast.est / Apo.Reg3.blast.html
Apo.Reg4.blast.est / Apo.Reg4.blast.html
- A continuació, es necessita passar els arxius txt resultat del MEGABLAST a format gff.
- Per fer això, cal exportar el programa PARSEBLAST, que és el que farà la transformació:
- En aquest moment, ja es disposa dels resultats de la comparació amb els ESTs en format gff per a cada regió, però aquests es troben en coordenades relatives.
- Per passar-ho altre cop a les coordenades absolutes inicials, és a dir, que el valor posicional de cada nucleòtid es correspongui a la posició real en la seqüència sencera. Cal sumar a l'inici i al final de cada regió el nombre de nucleòtids necessari.
- En aquest moment, ja es disposa dels resultats de la comparació amb els ESTs en format gff per a cada regió, però aquests es troben en coordenades relatives.
- Per passar-ho altre cop a les coordenades absolutes inicials, és a dir, que el valor posicional de cada nucleòtid es correspongui a la posició real en la seqüència sencera. Cal sumar a l'inici i al final de cada regió el nombre de nucleòtids necessari.
- Això s'ha realitzat a partir del següent programa en llenguatge gawk:
- Amb la resta de regions es fa de la mateixa manera:
- El que cal a continuació és eliminar els ESTs no rellevants o menys conservats per tal de quedar-nos amb els ESTs que realment són fiables per validar els gens predits.
- Observant els ESTs i els exons amb els quals coincideixen en els diferents gens, es podrà acceptar o rebutjar algun dels gens predits per als diferents programes.
-Mitjançant el script següent, es filtren només als ESTs que apareixen més d'un cop:
BEGIN{
OFS="\t";
}
{
nhsp[$9]++;
hsp[$9,nhsp[$9]]=$0;
}
END{
for (i in nhsp)
if (nhsp[i]>1)
for (j=1;j<=nhsp[i];j++)
print hsp[i,j];
}' Apo.Reg(X).blast.est.gff.absolut > SplicedEST.Reg(X).gff
gawk '
-Així doncs,al realitzar aquest programa per a cada regió, s'aconsegueix tenir arxius tipus SplicedEST.Reg(x).gff ( X és el nombre que indica la regió) on hi ha els hsp que ha triat el programa anterior.
- Abans però, s'ha comprovat quants ESTs (fragment de cDNA que s'expressa en un determinat teixit o moment del desenvolupament) hi havien repetits per a cada regió per corroborar que era bona, ja que no és convenient que hi hagi ESTs que només estiguin un cop, perquè no serien fiables per recolzar la predicció gènica. Podrien ser artefactes, és a dir, fragments de cDNA que s'han aliniat amb aquell exó però de manera inespecífica.
- El fet de trobar molts ESTs amb un mateix identificador és un bon senyal, ja que això implica que un mateix ESTs s'ha unit a diferents regions d'un gen predit (en diferents exons), és a dir, un fragment d'un EST s'uniria amb una zona d'un exó determinat, i el mateix EST es podria unir amb un altre exó del mateix gen predit.
- El que es pretén dir amb això, és que el trobar un mateix EST aliniat en diferents regions del genoma és un fort indicador que aquella és una zona d'splicing i, per tant, és molt probable l'existència d'un gen.
- Es va veure mitjançant la comanda següent que en gairebé totes les regions, els ESTs es trobaven força repetits, això fa preveure que la validació serà bastant fiable.
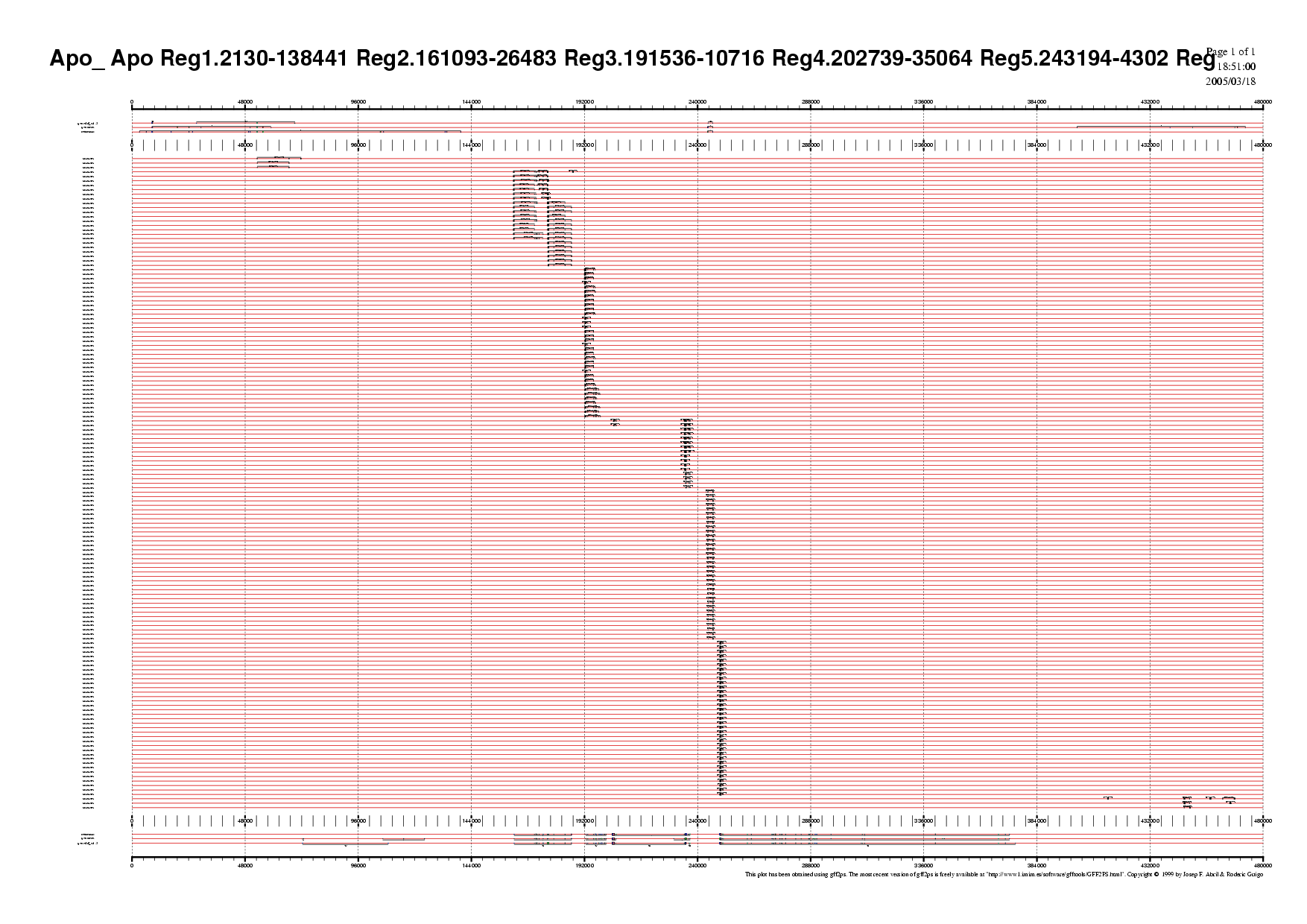
- Tot seguit, es va visualitzar la distribució dels ESTs juntament amb les prediccions gèniques dels 3 programes, per a cada regió de la seqüència, per corroborar quins són els ESTs que recolzen més les prediccions.
- Per altra banda, es vol una representació on no s'especifiqui si els ESTs són forward o reverse en el marc de lectura, per poder interpretar més còmodament els resultats a simple vista, ja que llavors els ESTs trobats apareixen al nivell mig de la imatge.
Per això, cal un programa en gawk que en comptes del frame afegeixi un punt. Aquest canvi es realitza a partir de l'arxiu gff.
Per poder obtenir aquesta imatge, com ja s'ha dit en apartats anteriors, cal transformar els arxius gff a ps i després a png, per visualitzar-los posteriorment amb el kview.
El procediment a seguir sería com el que es mostra a continuació per la regió 1:
- Per treure el frame:
- Per passar de gff a ps el que es fa és definir les coordenades de la regió en questió.
- Ara es passa a png, jugant amb la ressolució (densitat)posant-li un valor de 150:
- Per últim, es visualitza la imatge amb el programa kview:
- La modificació del frame es fa igual per a les altres 6 regions que falten,i es passa a ps definint els límits concrets de cada regió (els nucleòtids entre els quals es troba).
- Per a la regió 5 es va haver de posar nombres rodons a les coordenades ampliant els límits, perquè si no el programa no acceptava coordenades massa concretes:
- La conversió a png en aquestes 6 regions es fa exactament com per a la regió 1.
- Finalment, s'obtenen els arxius següents que contenen conjuntament els ESTs i gens predits per a regions determinades:
prediccions2.EST.Reg1.noframe.png
prediccions2.EST.Reg2.noframe.png
prediccions2.EST.Reg3.noframe.png
prediccions2.EST.Reg4.noframe.png
prediccions2.EST.Reg5.noframe.png
prediccions2.EST.Reg6.noframe.png
prediccions2.EST.Reg7.noframe.png
A partir de l'esquema obtingut, i seguint un criteri determinat, decidim quines prediccions dels gens són mé bones, és a dir, s'aproximen méa gens coneguts.
També s'ha fet una imatge en la qual es veu tota la seqüència ENm003 amb tots els ESTs trobats pel MEGABLAST i totes les prediccions de gen, i s'ha fet de la manera que prosegueix::
El que es va fer a continuació va ser mirar els ESTs que es van aliniar en la regió concreta (es va mirar per a les 7 regions), i es detecta si just en aquella zona on hi ha l'EST hi ha un exó predit per algun dels programes.
Com més ESTs hi hagi que concorden amb exons predits, més fàcil és acceptar aquell gen predit com a vàlid. Sobretot si els ESTs es troben en la regió 3' dels nostres gens.
Regió 1
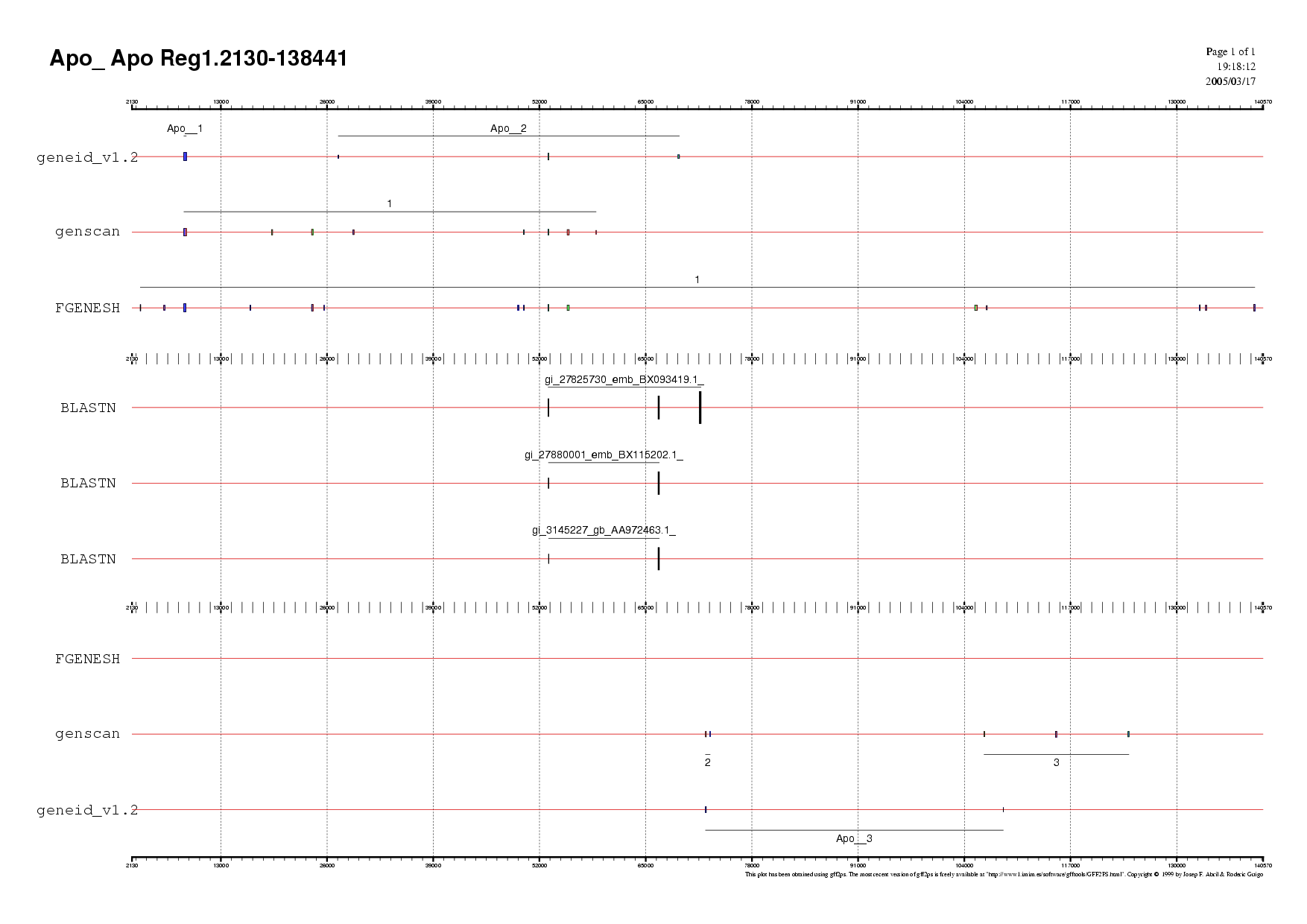
En aquesta regió, es va escollir com a fiable el gen 1 predit per fgenesh, perquè compartia més exons amb la predicció realitzada per Genscan que no pas amb la predita per Geneid, la qual vam descartar.
Les opcions estaven entre el gen 1 de genscan i el gen 1 de fgenesh. El d'elecció va ser el de fgenesh, tot i que va haver dubtes entre aquest i el de genscan, perqué en anar a buscar la seqüència aminoacídica, es va observar que la que donava genscan no començava per Met i, per tant, no es podria correspondre amb cap inici de prote&uiml;na.
Dels gens predits en reverse no ens quedem amb cap ja que no es pot aplicar cap mena de criteri per descartar o acceptar algun, donat que en cap ocasió hi ha recolzament per ESTs i, a més,les estructures exòniques no es correponen amb cap predicció ( cap exó igualment predit).
La seqüència del gen escollit és Gen 1
Regió 2
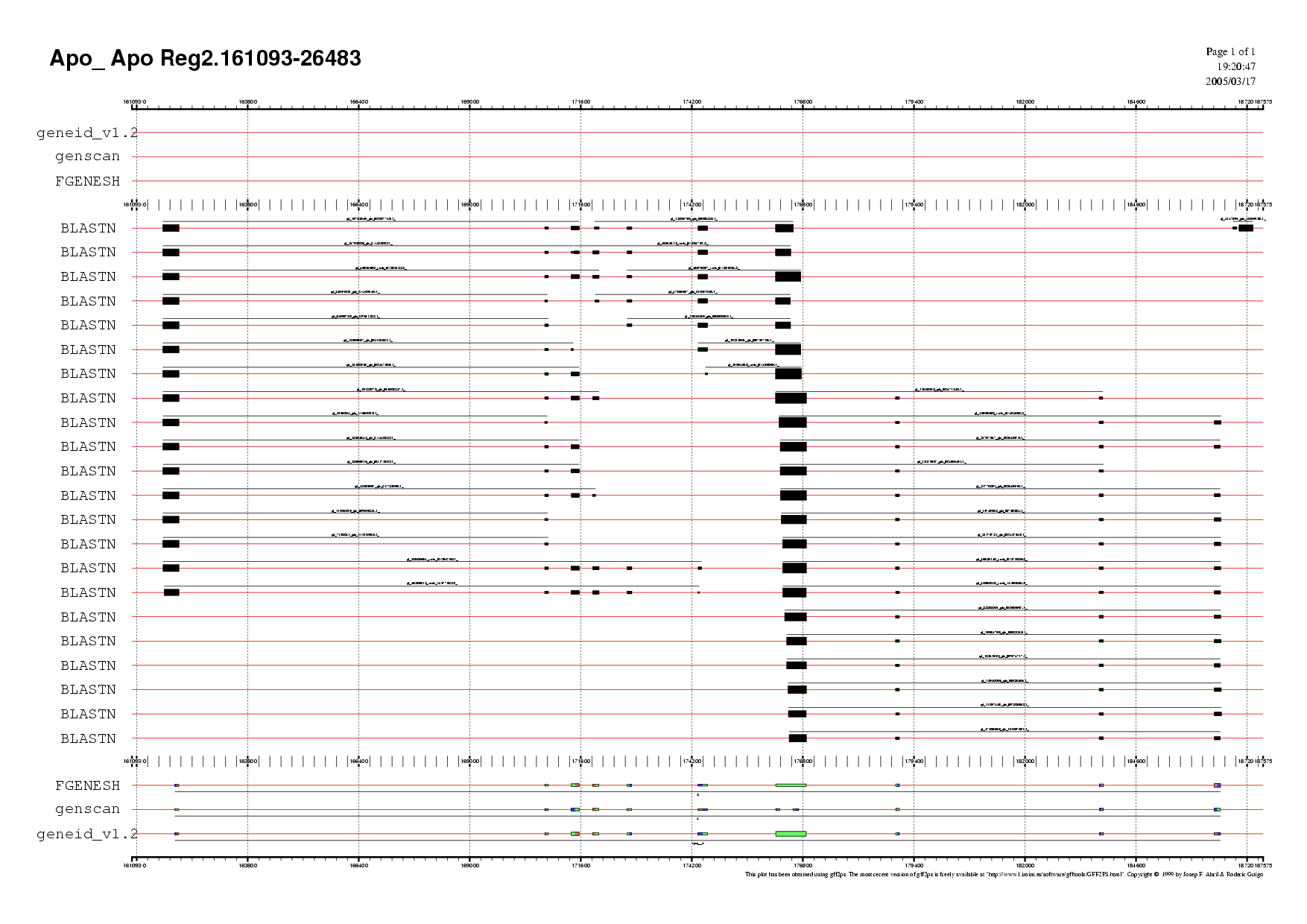
En la regió 2 hi havia un únic gen, predit pels 3 programes.
El gen de genscan va ser descartat perquè, tot i ser el mateix gen que el predit pels altres programes, hi havia un exó que no es veia recolzat per les altres prediccions.
Els gens de geneid i fgenesh eren completament idèntics i cada exó estava recolzat per ESTs. Finalment es va escollir el gen del programa fgenesh (reverse) perquè, encara que la predicció era igual de bona amb la de geneid (només es diferenciàven en un aminoàid), el programa fgenesh globalment havia permès acceptar més gens, i això fa pensar que potser aquest programa és més exacte que no pas geneid.
El gen escollit és el Gen 2
Regió 3
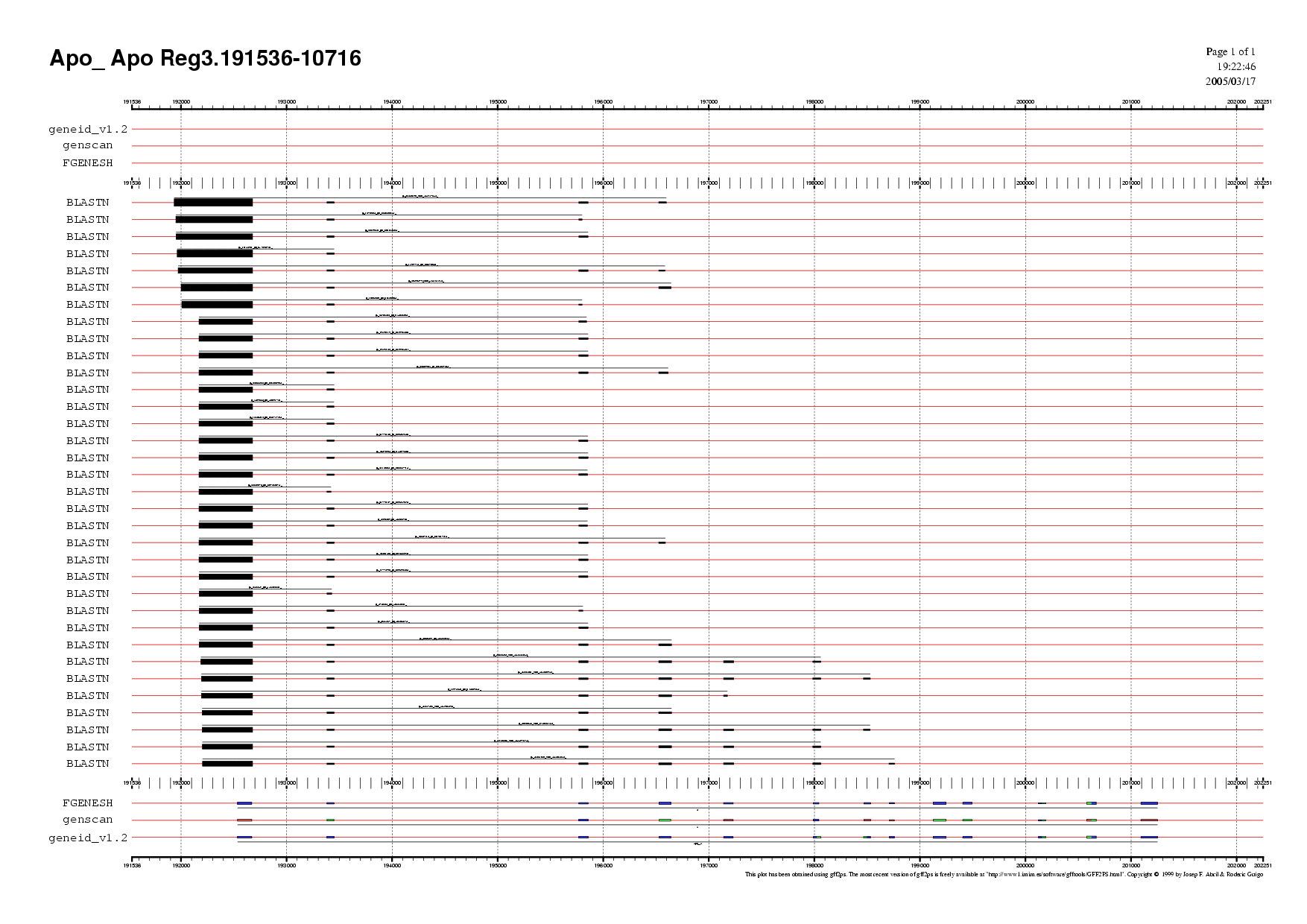
En aquesta ocasió tots els programes van predir el mateix gen, amb els mateixos exons, i molts d'ells suportats per ESTs.
Es va escollir el gen predit per geneid (reverse), tot i ser pràcticament idèntic al predit per fgenesh i genscan. El perquè de l'elecció radica en el fet que la predicció de geneid proporciona uns pocs aminoàcids més (6 exactament) que el programa fgenesh.
Això poder dòna una major possibilitat d'aliniament i, per tant, una major probabilitat de trobar alguna possible prote&uiml;na.
El gen de genscan ha estat descartat tot i tenir la mateixa predicció, ja que la pauta de lectura que ens ofereix és diferent a la dels altres dos programes.
El gen escollit correspon al gen 5 predit per geneid i ha estat anomenat Gen 3
Regió 4
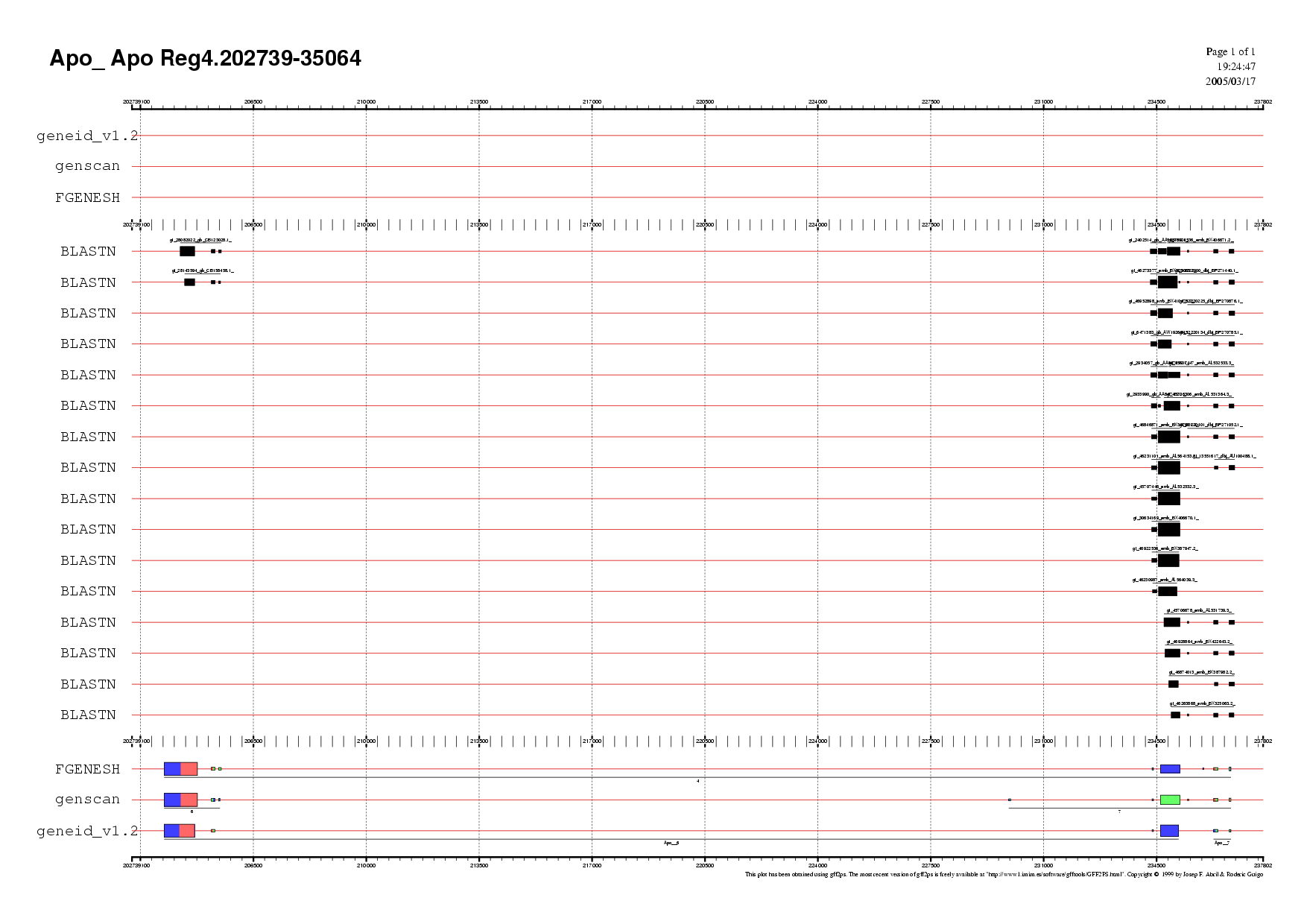
Es van descartar el gens predits per genscan ja que hi ha una zona que no contempla, mentre que les altres dues prediccions si que ho fan.
La decisió es trobava, doncs, entre escollir l'únic gen que prediu fgenesh o els dos que prediu geneid ( un d'ells era molt petit i es corresponia amb l'extrem 5' del gen de fgenesh).
El raonament seguit va ser el que prosegueix:
El gen escollit és anomenat Gen 4
Regió 5
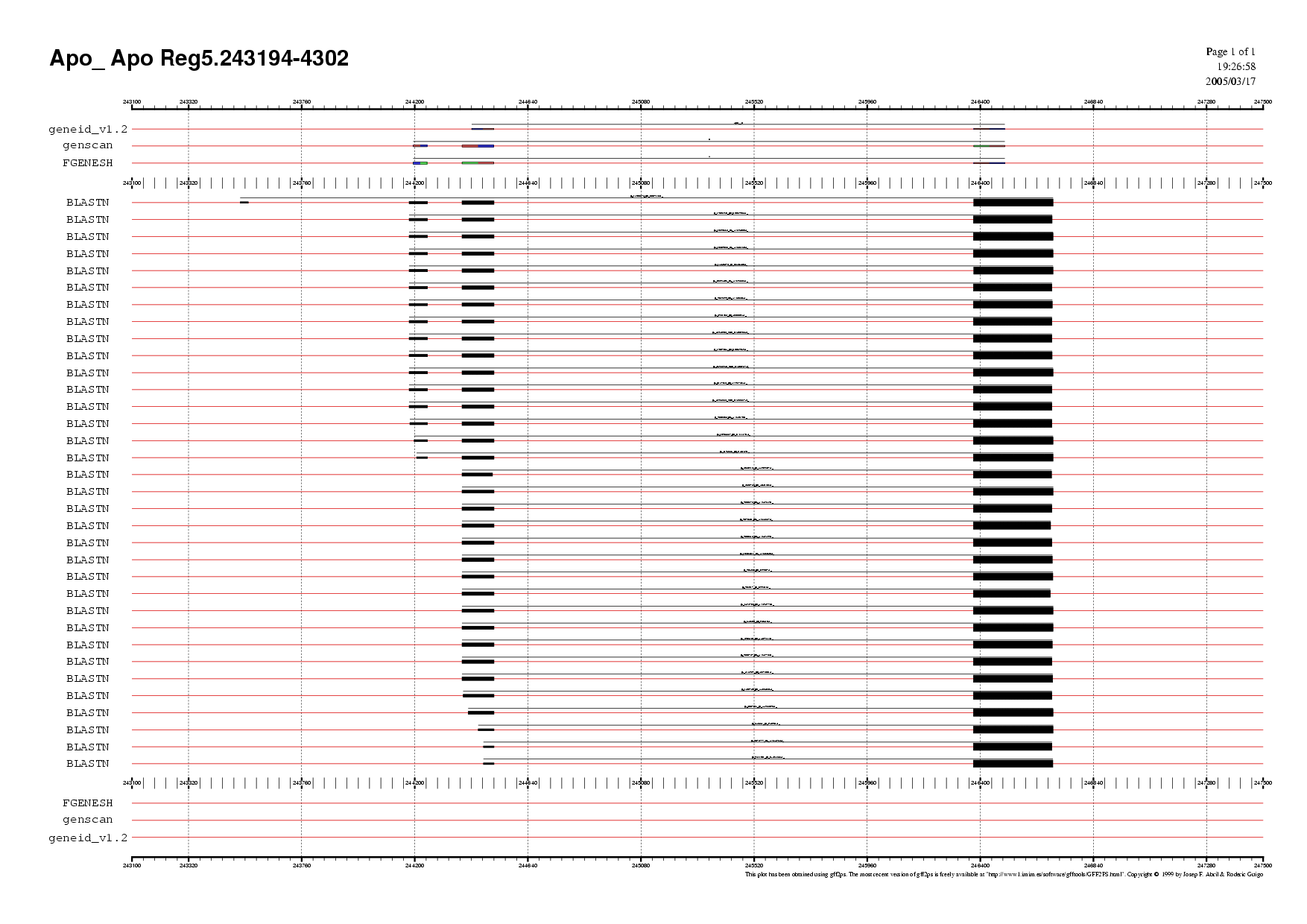
En aquesta regió es va escollir el gen que proporciona Fgenesh, perquè tot i que és el mateix que prediu genscan, en aquest últim es tenen evidències de que els marcs de lectura no es corresponen.
Geneid també va ser descartat, perquè el que prediu é diferent del que prediuen els altres dos programes.
El gen escollit té la seq%uuml;ència que es mostra en Gen 5
Regió 6
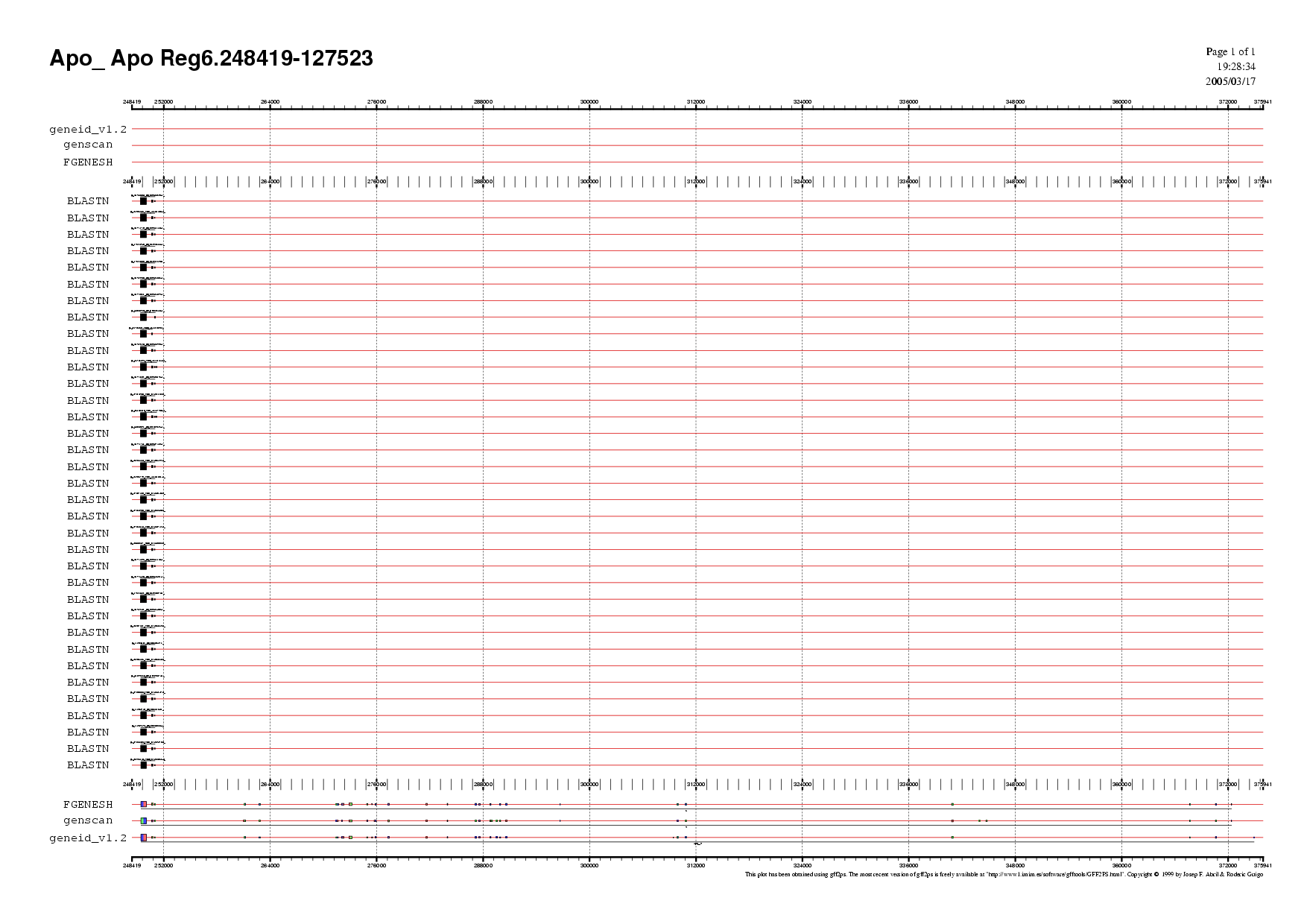
En la regió 6, s'ha agafat com a gen de referència el que prediu Fgenesh ( orientat en reverse), ja que tot i ser pràcticament igual als que es prediuen en els altres dos programes, es tenen evidències (pels gens que s'han escollit anteriorment), que Fgenesh proporciona millors prediccions.
El gen escollit és el s'ha anomenat Gen 6
Regió 7
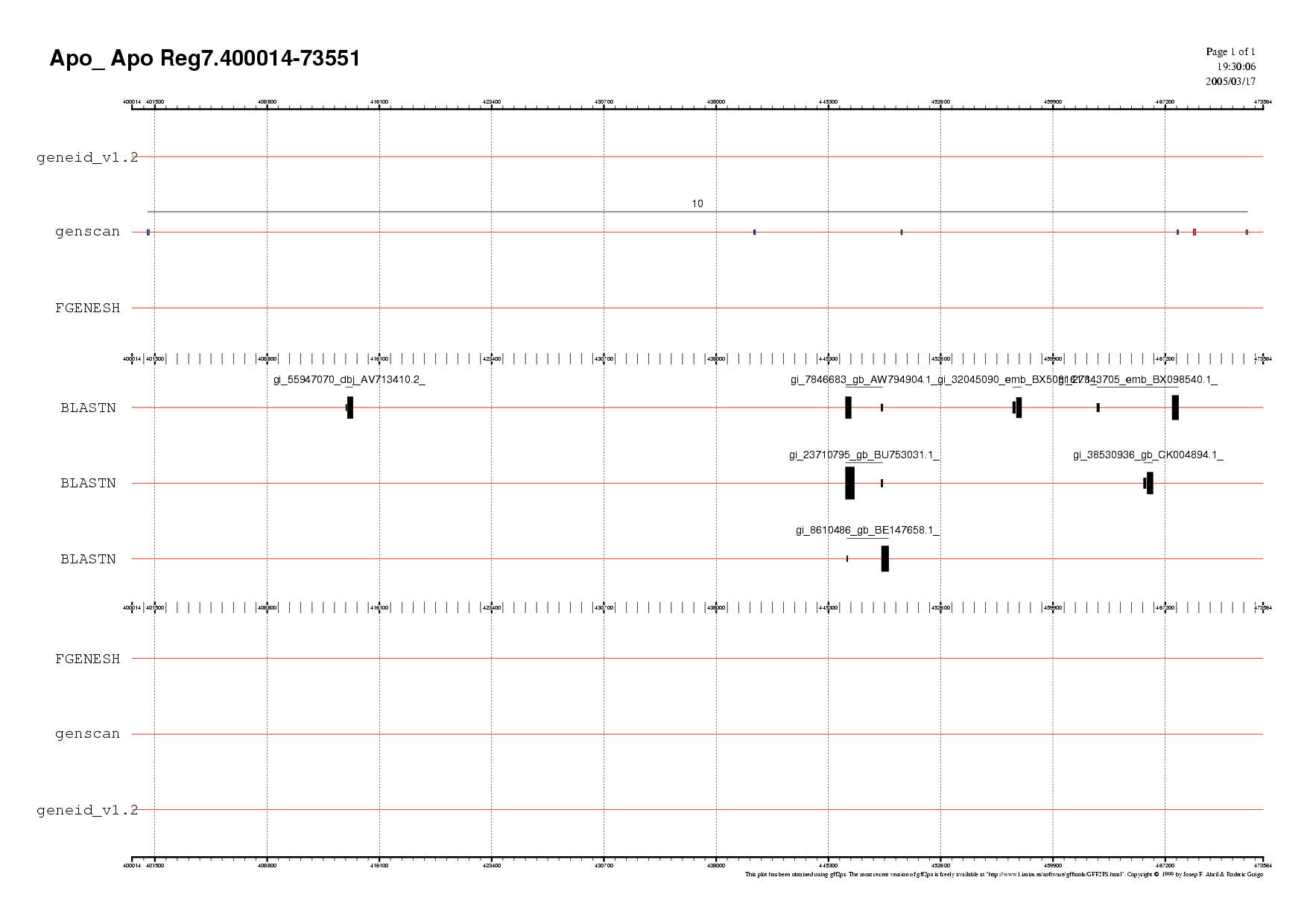
Per a la regió 7 s'ha considerat fiable el gen 10 predit per genscan (forward), ja que era l'únic gen predit en aquesta zona, i només per aquest programa.
És per aquest motiu que és interessant veure la seva homologia tot i no estar recolzat per cap EST.
La seqüència del Gen 7