
| Home ~ Intro ~ Project ~ Script ~ Results ~ Conclusions ~ Links |
SNPs distribution in genes Last Update: 21 March 2002 |
First of all, as the program consumes a lot of memory, it is better to split the origin data files intro chromosome specific ones. To do it the commands used from the shell are:
bash$ gawk '{ print $0 > "chroms/"$1".gff" }' refseq.gff
bash$ gawk '{ print $0 > "snps/"$2".txt" }' snpNih.txt
bash$ ls -1 chroms/chr* | \
perl -ne 'm/^.*chr(.*)\.gff$/o && print "$1\n";' > chrlist.tbl
bash$ cat chrlist.tbl | while read chr ;
do {
if [ -e snps/chr${chr}.txt -a -e chroms/chr${chr}.gff ];
then
echo "### Executing Localitzador at chromosome: $chr" 1>&2 ;
../localitzador snps/chr${chr}.txt chroms/chr${chr}.gff \
> output/chr${chr}.tbl 2> output/chr${chr}.err ;
else
echo "### ERROR!!! Check files of chromosome: $chr" 1>&2 ;
fi;
} ;
done ;
A complete readme-file is also avalaible.
To run the program you should type at the shell:
bash$ localitzador snps.txt exon.gff > snp_seq.tbl 2> snp_seq.err
The body of the program consists of 8 routines that drive the flow of the program. If you want you can see at first the complete version of the program.
We will explain all of them in the successive steps.
It verifies that the number of arguments at the first command-line is correct.
First of all it checks that the file referred by the first argument can be openned.
Afterwards it reads the SNPs file and counts the number of lines, storing it in a temporal vector (@f). Then it stores in a hash called %SNP{$snpchr} the fields number 2, 3, 4 and 5 of the line that is being read. This fields are:
This is a simple hash and its key is the number of chromosome.
It works in an analogous way like the last routine, but it checks the file referred by the second argument. Besides, the hash called %COORD{$chr}{$geneid} has an additional key that identificates the gene. Its structure, thereby, it is more complicated. This hash also has another fields:
At the @chr vector there are stored all the chromosomes read in the %SNP and this allows that with only one reference each vector in the hash can be accessed.
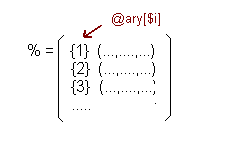
sub sort_snps() {
my @chr = keys %SNPS;
my $i = 0;
print STDERR "### SORTING SNPS DATA\n";
while ($i < scalar(@chr)) {
my $ary = \@{ $SNPS{$chr[$i]} };
print STDERR "# Chromosome $chr[$i] : sorting ".
scalar(@{ $ary })." SNPs...\n";
With the "map" function we may sort the acceptor and donor sites (stored as $snpstart and $snpend) from larger to smaller (indicated by "a" and "b") without creating a new hash, it just modifies the existing one (@{$ary} here). Then we assign to the mapping result the third position of the map vector ([2]).
if (scalar(@{ $ary }) == 1) { $i = $i + 1; next; };
@{ $ary } = map { $_->[2] }
sort { $a->[0] <=> $b->[0] # ordenem per acceptor
||
$a->[1] <=> $b->[1] } # ordenem per donor
map { [ $_->[0], $_->[1], $_ ] } @{ $ary };
$i = $i + 1;
};
}
It follows the same path but we need a previous step to access to the inner part of the data structure.
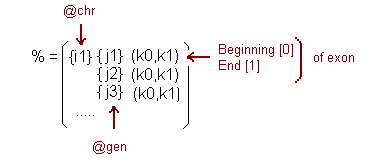
sub sort_seqs() {
my @chr = keys %COORDS;
my $i = 0;
print STDERR "### SORTING GENOMIC DATA\n";
while ($i < scalar(@chr)) {
my $ary = \%{ $COORDS{$chr[$i]} };
my @gen = keys %{ $ary };
my $j = 0;
print STDERR "# Chromosome $chr[$i] : sorting ".
scalar(@gen)." genes...\n";
while ($j < scalar(@gen)) {
my $arry = \@{ $ary->{$gen[$j]} };
if (scalar(@{ $arry }) == 1) { $j = $j + 1; next; };
@{ $arry } = map { $_->[2] }
sort { $a->[0] <=> $b->[0] # ordenem per acceptor
||
$a->[1] <=> $b->[1] } # ordenem per donor
map { [ $_->[0], $_->[1], $_ ] } @{ $arry };
$j = $j + 1;
};
$i = $i + 1;
};
}
To analyze this data we have to point at the variable of interest in the hash %COORDS and we use different alias (like $ary, $arry...) to refer each position.
sub analyze_data() {
my @chr = keys %COORDS;
my $i = 0;
print STDERR "### COMPARING SNPs COORDS AGAINST GENOMIC COORDS\n";
while ($i < scalar(@chr)) {
my $ary = \%{ $COORDS{$chr[$i]} };
my @gen = keys %{ $ary };
my $j = 0;
my $chrid = $chr[$i];
my $c = 0;
print STDERR "# Analyzing chromosome $chrid (";
scalar(@gen)." genes)\n";
while ($j < scalar(@gen)) {
my $arry = \@{ $ary->{$gen[$j]} };
my $k = 0;
my $genid = $gen[$j];
my $exon_num = scalar(@{ $arry });
while ($k < $exon_num) {
my ($ex_ini,$ex_end);
&counter(++$c);
$ex_ini = $arry->[$k][0];
$ex_end = $arry->[$k][1];
Then we use another routine, called checkcoords in order to calculate boxes that define three situations:
if ($exon_num == 1) {
&checkcoords($chrid,$genid,$ex_ini,$ex_end,'exon');
last;
};
if ($k > 0) {
&checkcoords($chrid,$genid,($arry->[$k-1][1] + $donor_len),
($ex_ini - $acceptor_len),'intron');
- Intern exon
- First or Last exon: because we do not want to calculate irreal acceptor sites in the first exon of a gene or donors in the last exon.
if ($k == 0) { # check first exon
&checkcoords($chrid,$genid,$ex_ini,
($ex_end - $donor_len),'exon');
&checkcoords($chrid,$genid,($ex_end - $donor_len),
($ex_end + $donor_len),'donor');
} elsif ($k == ($exon_num - 1)) { # check last exon
&checkcoords($chrid,$genid,($ex_ini + $acceptor_len),
$ex_end,'exon');
&checkcoords($chrid,$genid,($ex_ini - $acceptor_len),
($ex_ini + $acceptor_len),'acceptor');
} else { # check internal exon
&checkcoords($chrid,$genid,($ex_ini + $acceptor_len),
($ex_end - $donor_len),'exon');
&checkcoords($chrid,$genid,($ex_ini - $acceptor_len),
($ex_ini + $acceptor_len),'acceptor');
&checkcoords($chrid,$genid,($ex_end - $donor_len),
($ex_end + $donor_len),'donor');
}; # if first/last/internal
$k = $k + 1;
}; # while $k
$j = $j + 1;
}; # while $j
&endcounter($c);
print STDERR "# Chromosome $chrid done: ".scalar(@gen).
" genes - $c exons were checked...\n";
$i = $i + 1;
}; # while $i
} # read_seqs
As you can see &checkcoords needs some parameters:
This routine gives back the number and identity of the SNPs that has found in the box being checked. To do it, firstly it compares start and end of the box and it only works, obviously, if the beginning is smaller than the end. If this cheking is passed it reviews all the SNPs stored before under the alias $ary:
By accessing this last hash with references and alias, this routine gives back the concerned stored values.

Both programs are based in the same idea but, while in the first script the checkcoords entry is unique due to the previous sort, here there is one for each checkcoords. This mades the program's flow more complicated.
In addition, our program prepares data to be processed by the statistics module while the commented above does not. For further information look at the specifications of our program.
Click here to view the complete script.
Moreover, we have developed two accesory programs:
We have to point out that the acceptors and donor sites have been defined using a lenght that can be modified in the original script, changing the numerical value of:
We have used this lenght in order to simplify the consensus splicing sites.
There is another interesting and useful module we have learnt: Dumper, which allows to get a snapshot of complex data structures.
print STDERR Data::Dumper->Dump([ \%SNPS, \%COORDS ],[ qw( *SNPS *COORDS ) ] );