
Methodology
The aim of our project is to predict and annotate Danionella dracula’s selenoproteins and the translational machinery required for their synthesis and the proteins involved in selenium metabolism in this context. Given the precision in its genome sequencing and protein annotation, and considering its phylogenetic proximity to Danionella dracula, we selected Danio rerio as reference organism to perform our homology-based study.
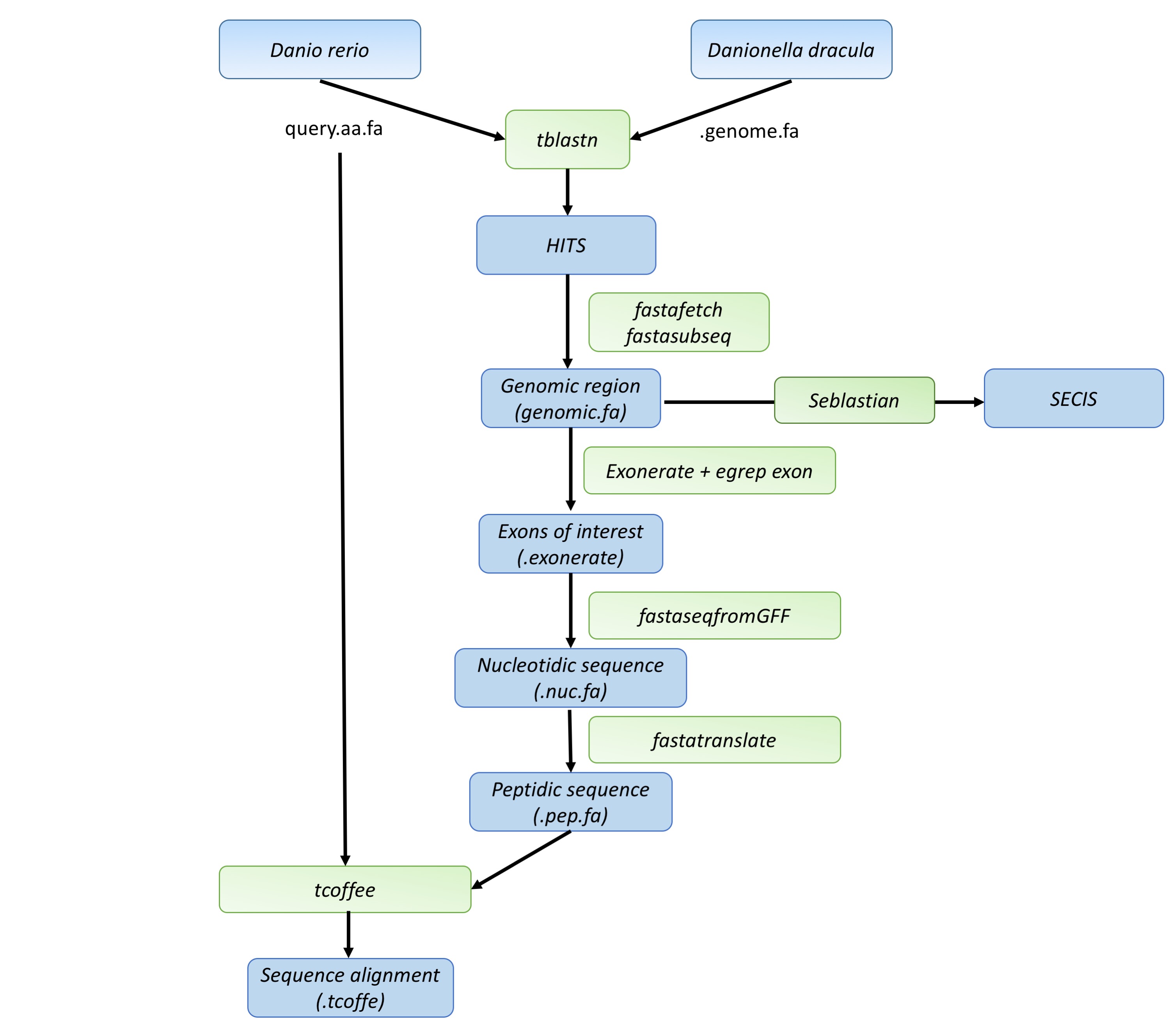
Workflow scheme of the project
Danionella dracula’s genome obtention
We obtained the Danionella dracula’s genome via a mounted directory provided by our Bioinformatics teachers. The genome was stored in the following directory:
/mnt/NFS_UPF/bioinfo/BI/genomes/2018/Danionella_dracula/genome.fa
Queries acquisition and processing
The used queries were originally from Danio rerio for the already described reasons. All queries were obtained from SelenoDB, the reference selenoprotein database. Queries were download as a multifasta (.mfa) file, and individually extracted using a python program made by us, which we link below. Selenocysteines appear as “U” when downloaded from SelenoDB, being unable to recognize by the programs we used. Therefore, “U” were changed for “X” , using our program, to carry on with the data extraction and analysis. All queries were saved as “query.aa.fa”.BLAST
The BLAST (Basic Local Alignment Search Tool) program works via algorithms to perform local alignments between our query and the reference Danio rerio’s genome. Given that our query was a proteic sequence we used tBLASTn, a BLAST variation that implements a proteic to nucleotidic sequence change before comparing the query to the reference genome.[14]
We based our hit selection on the E-value, only taking into account hits with E-values higher than 0.01, to discard haphazard alignments. All blasts executed and stored as “query.blast” files before moving on into the next steps of our methodology.
os.system("tblastn -outfmt 6 -query %s -db %s -out %s -evalue 0.01" % (query,genome,out_blast))
Fastafetch
Once we selected the desired hits, Fastafetch command allows us to extract the scaffold in which our hit is contained. The indexed genome was also provided by our bioinformatics teachers in the following directory:
/mnt/NFS_UPF/bioinfo/BI/genomes/2018/Danionella_dracula/genome.index
os.system(“fastafetch %s %s %s > ./fastafetch/%s.fa" %(genoma, genomaindex, scaffold_id, query))
Fastasubseq
The fastasubseq command permits the extraction of our genomic region of interest inside the scaffold. Since blast only provides hits in exons we extended 50.000 nucleotides in each side so that the presence of introns won’t result in the omission of coding sequence. In those cases where the 50.000 nucleotide extension went beyond scaffold 3’ and 5’ limits our program reset the extension limits to the start or end of the scaffolds respectively.
os.system("fastasubseq ./fastafetch/%s.fa %i %i > ./fastasubseq/%s_genomic.fa" %(query,inici,length,query))
Exonerate
Exonerate extracts the predicted gene for our genomic region of interest. To extract just the exons we used the egrep command.[14]
os.system(“exonerate -m p2g --showtargetgff -q ./proteins/%s.aa.fa -t ./fastasubseq/%s_genomic.fa | egrep -w exon >
FastaseqfromGFF
This function generates the cDNA sequence corresponding to the exonerate gene prediction results. In order to use it it may be necessary the exportation of the program, which was implemented at the start of our program using our function module containing the following export path:
export PATH=/mnt/NFS_UPF/bioinfo/BI/bin:$PATH
os.system("fastaseqfromGFF.pl ./fastasubseq/%s_genomic.fa ./exonerate/%s.exonerate.gff > ./fastaseqfromGFF/%s.nuc.fa"%(query,query,query))
Fasta translate
This program translates the cDNA protein generated by fastaseqfromGFF into protein sequence. In order to perform a correct t-coffee alignment we must change the selenocysteines translated as “*” into “X”, this step was also automatized in our program. The F1 translates only the first ORF.
os.system("fastatranslate -F 1 ./fastaseqfromGFF/%s.nuc.fa > ./fastatranslate/%s.pep.fa"%(query,query))
T-coffee
T-coffee generated global alignment between our initial Danio rerio’s query and the predicted protein for Danionella dracula.[16]
os.system("t_coffee ./proteins/%s.aa.fa ./fastatranslate/%s.pep.fa > ./tcoffee/%s.tcoffee" %(query,query,query))
SEBLASTIAN
Seblastian allowed us to perform an in silico SECIS element search. As mentioned before SECIS elements are found in the 3’- UTR of selenoproteins and are fundamental for selenocysteine synthesis. Seblastian results were contrasted and complemented with data extracted from the just explained process above. [17]Phylogenetic trees
In order to assess the predictions phylogenetic trees were generated using phylogeny.fr software. Inputs were submitted as multifasta files containing the Danio rerio’s query and the predicted sequences. [18]Program
Program codeProgram
Function Module code