Genomes selection
The philogenetically closest species to Nannopterum harrisi is Taeniopygia guttata. However, this is not a very well studied organism. Thus, we decided to compare our species also with Gallus gallus, a model animal, much better characterized.
The genome of our species was provided in a multifasta file by the Pompeu Fabra University in the following directory:
The sequence of Gallus gallus and Taeniopygia guttata was obtained from selenoDB 2.0, also in a multifasta file.
Procedure
FROM MULTIFASTA TO FASTA
First of all, we had to convert our multifasta file with all selenoprotein we had obtained in the selenoDB to different single fasta file to obtain every protein separated.
CHANGE U FOR X
Selenoproteins contain the selenocystein aminoacid (U) which is not a conventional aminoacid, thus it is usually misread in some of the programs used, such as Exonerate, T-Coffee and GeneWise. Besides, some of our fasta files contained strange symbols such as @ or # or % so we decided to eliminate them.
In order to do so we used the next command:
$sed 's/[#@%]//g' ./selenoproteines/$nomfitxer/$nomfitxer.ucanviat.fa > ./selenoproteines/$nomfitxer/$nomfitxer.canviat.fa
BLAST
In order to find selenoproteins in Nannopterum harrisi, we performed a Blast, that confronted our genome with the sequences of the already annotated genome of T. guttata and G. gallus. Blast uses an heuristic algorithm to compare sequences, find similarities and finally to obtain statistical parameters.
In our project, we only wanted significant hits. Thus, we selected only the alignments with an e-value lower than 0,01 to make sure this hits were not randomly produced and had statistically significant meaning.
We used tblastn as we wanted to compare a sequence of aminoacids of the query with sequence of nucleotides of N. harrisi genome. As for the output format, we used format 6.
The command used in our program was:
/genome.fa -evalue 0.01 -outfmt 6 -out ./selenoproteines/$protein/$protein.canviat.blast
In which the path after -query is referring to the fasta file of either T. guttata or G. gallus and the path after -db is referring to the sequence of nucleotides of Nannopterum harrisi. At the end of the command, there is specified the path in which the output file is going to be saved (-out).
The output file of the blast is a list of the scaffolds, with their locations, the identity and the e-value of every hit. Because tblastn always sorts the scaffolds taking into account the best e-value, our program automatically chooses the scaffold in the first hit as the best one and proceeds with this one.
However, we also need to consider duplications, meaning that there is a probability that more than one scaffold is statistically significant in every protein. As read in the literature, in birds, in some cases duplications have been found. Thus, we wrote a second program which read every output file of tblastn, show it in the terminal allow us to choose, if necessary a second scaffold, and finally doing the rest of the automatization with it (in order to obtain this program click here).
OBTENTION OF THE REGION OF INTEREST
After having selected our hit, we needed to extract the region of interest in from the both the Gallus gallus or Taeniopygia guttata genome. In order to do so, the scaffold containing the region was extracted using fastafetch. The fastafetch command extracts a given sequence or scaffold from an input file and an indexed file.
In our script, this action corresponds to the following command:
/cursos/20428/BI/genomes/2017/Nannopterum_harrisi/genome.index '$contig' > './selenoproteines/$protein/$contig.fa'
Once the scaffold was selected, the command fastasubseq allowed us to obtain a smaller region of interest within this contig. In order to do so, we extended the area of our hit 50.000 nucleotides per site. The following command was used in our program:
GENE PREDICTION
In order to predict the genes within our query sequence we used exonerate. Exonerate aligns and compares the sequence obtained by fastasubseq with the DNA sequence of the query, predicting the coding regions, the exons, and the non-coding regions and the introns.
We used the following command, in which we decided to to include the exhaustive prediction for a better analysis:
Once the exonerate was done, we used T-Coffee to to compare the predicted proteins of N. harrisi with the query of the T. guttata or G. gallus. In order to do so, we needed to predict the protein of N. harrisi from the output file of the exonerate. First, we obtained the exon parts of the gene predicted using the following command:
'./selenoproteines/$protein/$contig/$protein.exon.gff'
Afterwards, once the coding sequence is obtained, we need to know the cDNA it is going to be generated considering the exons. In order to do so, we used the following command:
'./selenoproteines/$protein/$contig/$protein.exon.gff' > './selenoproteines/$protein/$contig/$protein.prednt.fa
Previously, we need to translate the nucleotide sequence previously obtained to aminoacids, in order to obtain the protein predicted. The following command was used:
'./selenoproteines/$protein/$contig/$protein.predaa.fa
One last step before the T-Coffee is to change the * for X in predicted protein.
'./selenoproteines/$protein/$contig/$protein.Xpredaa.fa
At last, we proceed with the T-Coffee command. This command allows us to determinate if the two proteins are homologous and if there is a highly conserved aminoacid sequence or not.
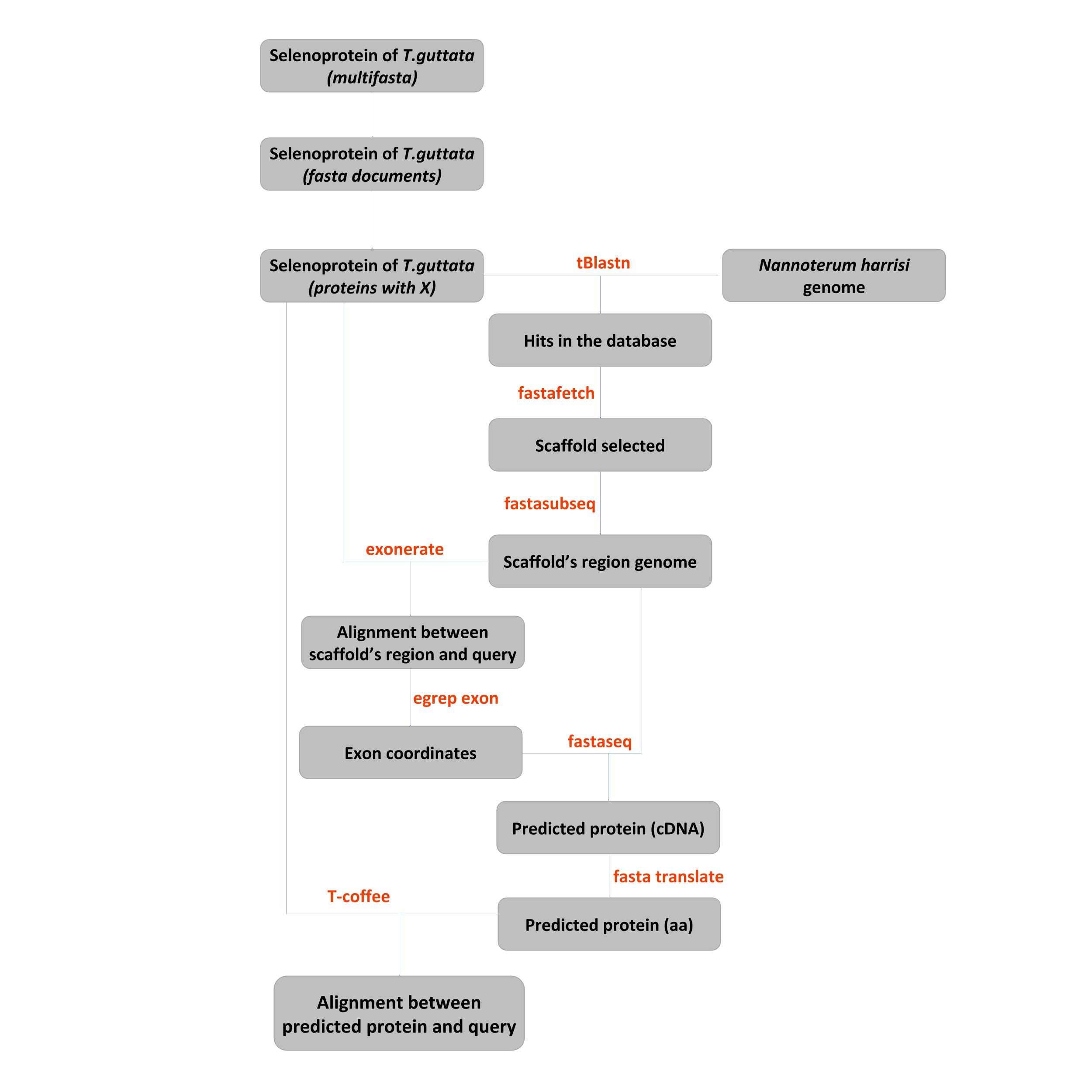
Automatization
In order to automatizate the previously explain process, an automatized program was used (to obtain the program click here).
SECIS elements
In order to make sure that the predicted proteins really are selenoproteins, SECISerch3/Seblastian was used. Its aim is to find SECIS elements in the fastasubseq output file (SECIS elements are structural motifs of 60 nucleotides located in the 3'-UTR of the genes encoding for a selenoprotein).