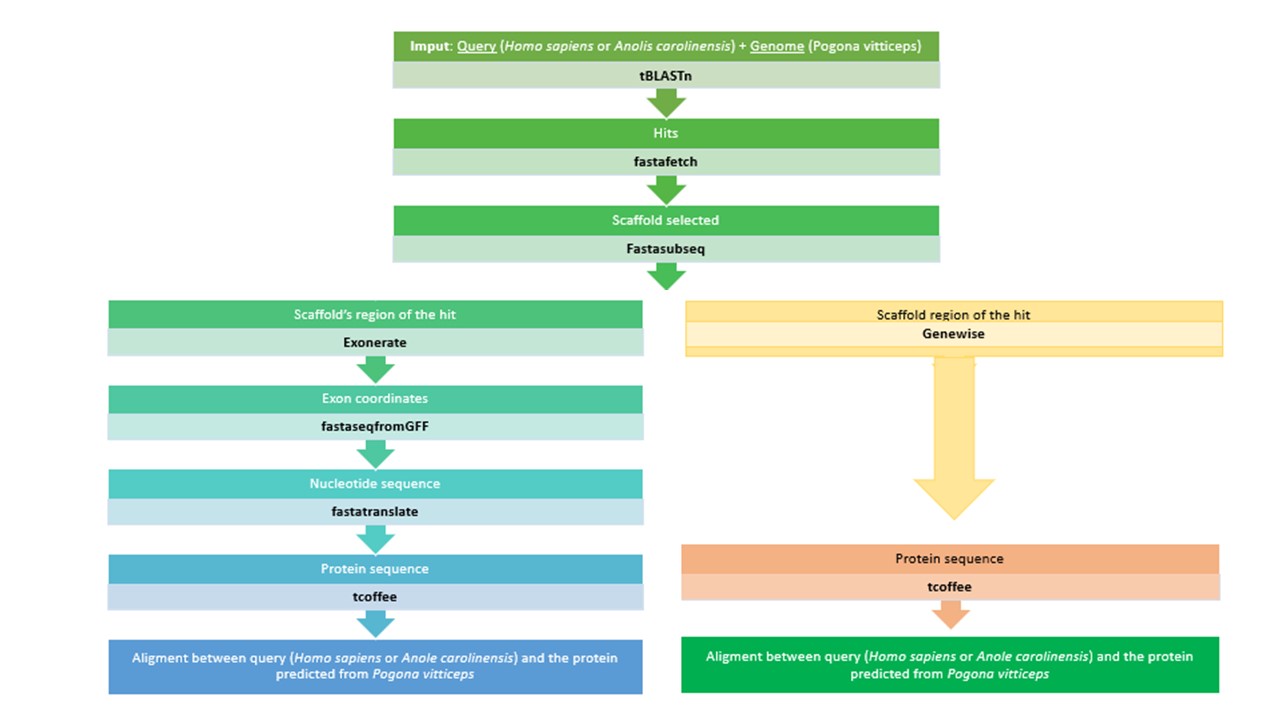
The aim of this study was to carry out a novel prediction of the selenoproteins and the respective machinery from the Pogona vitticeps genome. We executed a homology based approach from the Anolis carolinensis and the Homo sapiens genome.
1. Obtention of the genome
The genome of study is the Pogona vitticeps genome. We downloaded it from a file created by the professors:
/cursos/20428/BI/genomes/2017/Pogona_vitticeps/genome.fa.
We were also provided with an indexed genome which we obtained from:
/cursos/20428/BI/genomes/2017/Pogona_vitticeps/genome.index.
2. Obtention of the queries
We have chosen the Anolis carolinensis genome as the reference genome to identify the selenoproteins because it is the closest specie to ours (to the Pogona vitticeps genome) with a well-annotated genome. The selenoproteins and the machinery proteins were obtained from SelenoDB database 2.0 in fasta format. Every protein was classified in a single file and was saved with the name of the protein.
We also obtained the human selenoproteins, in this case form the SelenoDB database 1.0 and they were classified in a single fasta and saved with the name of the protein plus “_human”.
3. Prediction process
We used a combination of automatic and manual procedure to predict the Selenoproteins. We created two programs in order to do that: the first one to do the blast and the other one to do the remaining steps.
tBLASTn
ANALYSIS
4. Loading modules
We started by loading the modules and the paths needed to execute all the programs. These were:
module load modulepath/goolf-1.7.20
module load BLAST+/2.2.30-goolf-1.7.20
module loadExonerate/2.2.0-goolf-1.7.20
module load T-Coffee/11.00.8cbe486-goolf-1.7.20
export PATH=/cursos/20428/BI/bin:$PATH
export PATH=/cursos/20428/BI/soft/Genewise/x86_64/bin:$PATHexport
WISECONFIGDIR=/cursos/20428/BI/soft/Genewise/x86_64/wise2.2.0/wisecfg/
5. Substitution of U to X
The sequences containing a selenocysteine (U) were modified replacing the U for an X as the bioinformatic resources used for the analysis do not recognize this character. The #,@ and other characters were also removed.
6. Comparing sequences: tBLASTn
tBLASTn allows us to find regions of similarity between sequences. The program compares proteins vs translated genomic sequences. tBLASTn provides us some values that we used for our analysis. The E-value, is the number of hits that are expected to be found by chance. Therefore, the lower E-values corresponds to a higher hit significance. In our case we took for further analysis those hits with an E-value < 0.05.
The command needed to execute this step is the following:
tblastn -query $selenoprotein -db /cursos/20428/BI/genomes/2017/Pogona_vitticeps/genome.fa -outfmt 7 -out selenoprotein.blast
7. Obtention of the regions of interest
The following step consists on extracting those parts of the genome of Pogona vitticeps that match with the protein of Anolis carolinensis or the Homo sapiens genome.
7.1. Isolation of the contig of interest: FASTAFETCH
We generated a file with the scaffold sequence by using the command:
fastafetch genome.fa genome.index ‘$scaffold’ > “$scaffold.fa”
Here, the input file is the database of Pogona vitticeps and the indexed file the one obtained by downloading it at the beginning as we have mentioned before.
7.2. Bounding the selenoprotein: FASTASUBSEQ
Then, we extracted a scaffold subsequence that had 100.000 base pairs upstream and 100.000 base pairs downstream the blast hit. The command used was:
fastasubseq $scaffold.fa start length > $query.$scaffold.genomic.fa
8. Gene prediction within every subsequence
Once the subsequence was obtained, the next step consisted on predicting the gene coordinates. It was done by using Exonerate and Genewise, genetic tools for pairwise sequence comparison. We created a file with all the predicted exon coordinates.
We launched the program with the following command:
Exonerate --exhaustive yes -m p2g --showtargetgff -q $query -t $query.$scaffold.genomic.fa | egrep -w exon > $query.$scaffold.Exonerate.gff
We decided to include the exhaustive prediction for a more precise analysis.
After that, an amino acidic sequence file was obtained with the Anolis carolinensis or Homo sapiens query protein. To do that, we first generated the nucleotide sequence and then the amino acidic sequence with the commands:
fastaseqfromGFF.pl $query.$scaffold.genomic.fa
$query.$scaffold.exonerate.gff > $quey.$scaffold.nt.fa
fastatranslate -f $query.$scaffold.nt.fa -F 1 > $query.$scaffold.p.fa
The "*" symbols that correspond to codon stops or selenocysteine codons were substituted by X. We finally run the T-Coffee in order to obtain the alignment between the Anole carolinensis or the Homo sapiens query and the predicted protein of Pogona vitticeps:
t_coffe $query $query.$scaffold.pX.fa > $query.$scaffold.tcoffe
We created an excel document where the results for each selenoprotein were annotated. For each hit with the E- value < 0.05 the following parameters were annotated:
- Gene location
- Number of exons
- Score
- Strand positive or negative
- Starting with methionine or not
- Alignment or not of X in query and database
- Alignment or not of C in query and database
The Genewise software was used to validate the Exonerate prediction that we’ve obtained for all the proteins. We executed the Genewise software with the query against the SUBSEQ sequence (file saved as “genomic.fa”) with the following commands:
Genewise -pep -pretty -cdna -gff -trev query protein.genomic.fa > protein.Genewise
The output obtained was saved with the name of the protein followed with the extension “.Genewise”
With the results obtained from the Genewise, we performed a t-coffee alignment with the query in order to compare this t-coffee output with the t-coffee obtained using the Exonerate software.
Figure 5. Summary of materials and methods.
9. SECIS and Seblastian search
We used the Seblastian software (link) as an additional tool for selenoprotein prediction. The input of the software was the file obtained for the fastasubseq. SECIS elements were predicted using the SeciSearch3 tool. The input for this software was also the file obtained for the fastasubseq.