MATERIALS I MÈTODES
L'objectiu del nostre treball és identificar les selenoproteïnes en el genoma de Python bivittatus . Per dur a terme aquest treball, hem comparat les selenoproteïnes ja anotades d’Anolis carolinensis i Homo sapiens amb el nostre genoma, mitjançant els programes que s'explicaran a continuació.
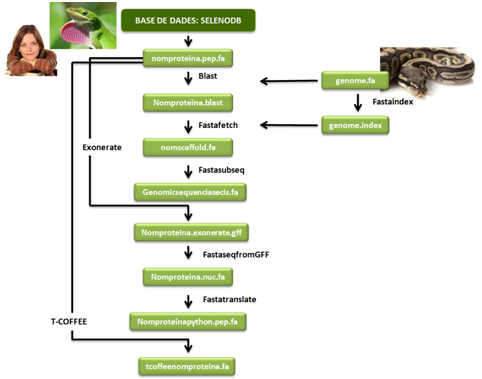
Obtenció del genoma de Python bivittatus
El genoma de Python bivittatus s’ha obtingut a partir d’una base de dades que ens han proporcionat els professors de l’assignatura. Per tal d’obtenir-lo s’ha hagut de recórrer al següent directori:
/cursos/BI/genomes/vertebrates/2014/Python_bivittatus/genome.fa
En aquest directori es pot trobar el genoma de Python bivittatus, però per a poder utlitlizar-lo durant el procés de recerca de les selenoproteïnes cal que s’indexïi (separar el genoma en segments i ordenar-lo en scaffolds). A continuació es mostra la comanda que s’utilitza per a indexar el genoma:
$ fastaindex /cursos/BI/genomes/vertebrates/2014/Python_bivittatus/genome.fa genome.index
El genoma indexat s’emmagatzema en un fitxer que s’anomena genome.index.
Obtenció de les seqüències queries
Amb l’objectiu d’obtenir les seqüències de les selenoproteïnes en Anolis carolinensis i Homo sapiens, s’ha utilitzat la base de dades SelenoDB.
Les selenoproteïnes d’Anolis carolinensis s’han utilitzat com a queries ja que el llangardaix Anolis és una espècie filogenèticament propera a Python bivittatus. Les seqüències corresponents a aquestes proteïnes s’han alineat amb el nostre genoma per saber si també es troben en la pitó. Tot i així, algunes selenoproteïnes d’Anolis carolinensis no es troben tan ben anotades com en el genoma d’Homo sapiens. En aquests casos, s’ha partit de selenoproteïnes humanes a l’hora d'estudiar-les en el genoma d’interès.
Una vegada s’han obtingut les seqüències d’àminoàcids de les selenoproteïnes, s’han emmagatzemat en un fitxer anomenat nomproteina.pep.fa (Ex. selm.pep.fa).
Abans de començar a executar comandes amb el terminal s’han de donar una sèrie de permisos que permeten exportar tots els programes necessaris per realitzar la recerca de selenoproteïnes. A continuació es presenten les comandes que s’han d’executar al terminal:
| fastaseqfromGFF.pl | $ export PATH=/cursos/BI/bin:$PATH |
| NCBI Blast | $ export PATH=/cursos/BI/bin/ncbiblast/bin:$PATH |
| NCBI Blast | $ cp /cursos/BI/bin/ncbiblast/.ncbirc ~/ |
| Exonerate | $ export PATH=/cursos/BI/soft/exonerate/i386/bin:$PATH |
| T_coffee | $ export PATH=/cursos/BI/soft/t_coffee/i386/bin:$PATH |
| GeneWise | $ export PATH=/cursos/BI/soft/genewise/i386/bin:$PATH |
| GeneWise | $ export WISECONFIGDIR=/cursos/BI/soft/genewise/i386/wise2.2.0/wisecfg/ |
Selecció de l'Scaffold
BLAST (Basic Local Alignment Search Tool) és un programa informàtic d’alineament de seqüències biològiques ja siguin nucleòtids o aminoàcids. El programa permet comparar una seqüència problema (query) contra una gran quantitat de seqüències que es trobin en una base de dades. El programa utilitza un algoritme heurístic que troba les seqüències o hits de la base de dades amb major homologia respecte la seqüència query. És important esmentar que, com que BLAST utilitza un algoritme heurístic, no es pot garantir que hagi trobat la solució correcta ja que pot comportar la pèrdua de hits reals que no presenten una gran similtud.
Existeixen diferents tipus de BLAST segons el format de les seqüències que es volen alinear. En el nostre cas, s’ha utilitzat el tBLASTn, el qual permet comparar una seqüència proteica (query) amb una base de dades de seqüències nucleotídiques (el genoma de Python bivittatus).
Per executar el programa es va utilitzar la següent comanda:
$ blastall -p tblastn -i fitxerquery.fa -d nombbddBLAST -o fitxerdesortida
-p és el tipus de blast utilitzat (tblastn), -i és el fitxer on es troba localitzada la query, -d és el genoma de Python bivittatus i -o és el nom del fitxer on es guarda el resultat del blast (output).
L’output del tblastn conté un llistat dels possibles alineaments entre la query i el genoma contra el qual s’ha alineat aquesta. Cadascun d’aquests alineaments és un hit amb un E-value determinat, que permet valorar la significança estadística de cada hit. Aquest paràmetre descriu el nombre de hits, és a dir, el nombre d’alineaments que es poden esperar únicament per atzar mitjançant l’anàlisi amb BLAST.
Cal remarcar que en cas de tenir múltiples hits, s’escullen els hits amb el millor E-value, major percentatge d’indentitat i millor score. Per altra banda, cal considerar que la selenocisteïna o alguna cisteïna de la query es pot trobar alineada amb un codó STOP, que indicaria una possible predicció d’una selenoproteïna, o amb una cisteïna, que indicaria un possible homòleg en cisteïna.
Extracció de la regió genòmica
Un cop s’han obtingut tots els hits i s’han triat els que es volen analitzar, cal extreure la regió genòmica on l’alineament amb el tblastn indicava que es podria trobar la proteïna estudiada. Com que anteriorment ja s’ha indexat el genoma, es pot executar directament el programa Fastafetch, que permet l’extracció dels scaffolds on es troba cadascun dels hits que s'han seleccionat. Per executar aquest programa es va utilitzar la següent comanda:
$ fastafetch cursos/BI/genomes/vertebrates/2014/Python_bivittatus/genome.fa genome.index nomscaffold > nomscaffold.fa
On nomscaffold es refereix al nom de l’scaffold seleccionat i nomscaffold.fa al fitxer que es crea quan executem aquesta comanda.
Seguidament, dins de l’scaffold seleccionat, es delimita encara més la regió d’interés per tal d’aconseguir seqüències més curtes i més precises del fragment que possiblement contingui la selenoproteïna. Amb l’objectiu d’extreure el gen sencer d’interès, s’agafen les posicions del hit obtingut al BLAST i s’expandeixen els marges downstream i upstream de l’alineament. La mida de l’expansió que es va realitzar va ser aproximadament de 50.000 nucleòtids upstream i 50.000 downstream. D’aquesta manera també es garanteix incloure l’element SECIS del gen. No obstant, aquesta mida pot variar segons la longitud i propietats de la query o de l'scaffold. Per tal d’acotar la regió s’utilitza el programa Fastasubseq mitjançant la següent comanda:
$ fastasubseq nomscaffold.fa start length > genomicsequenciasecis.fa
En aquesta comanda s’introdueix l’scaffold seleccionat (nomscaffold.fa), el nucleòtid per on comença la nostra regió (start) i la longitud de nucleòtids que contindrà aquesta (lenght). La regió seleccionada es guardarà en un fitxer de sortida anomenat genomicsequenciasecis.fa en el qual “seqüència” fa referència al nom de la selenoproteïna que s’està analitzant.
Predicció del gen
Exonerate
Un cop s’ha extret el fragment de DNA que conté el hit d’interès mitjançant el Fastasubseq, cal assegurar que el hit es troba dins l’exó, i per tant, codifica per una proteïna. Exonerate és un programa que permet predir l’estructura gènica de la seqüència problema en el genoma de Python bivittatus. La comanda per executar aquest programa és la següent:
$ exonerate -m p2g --showtargetgff -q nomsequencia.pep.fa -t genomicsequenciasecis.fa --exhaustive yes
on -m indica el model d’alineament, en aquest cas p2g (protein to genome), és a dir, compara la query contra el genoma. El paràmetre -q indica on es troba el fitxer amb la query i -t, la seqüència contra la que es compara la query, és a dir, la regió de l'scaffold en la qual es creu que hi ha el gen d'interès. Finalment, --showtargetgff emmagatzema el fitxer de sortida en format GFF.
A l’hora d’executar la comanda és important canviar, en el fitxer on està la query, les possibles “U” per “X” ja que Exonerate no reconeix la lletra U i donaria FATAL ERROR.
Per tal d’obtenir només els exons s’utiliza la comanda egrep -w exon:
$ exonerate -m p2g --showtargetgff -q sequencia.pep.fa -t genomicsequenciasecis.fa --exhaustive yes | egrep -w exon >
sequencia.exonerate.gff
FastaseqfromGFF
Seguidament, s’ha utilitzat el programa FastaseqfromGFF.pl, que extreurà la seqüència exònica en format FASTA mitjançant la comanda:
$ fastaseqfromGFF.pl genomicsequenciasecis.fa sequencia.exonerate.gff > sequencia.nuc.fa
on genomicsequenciasecis.fa és la subseqüència que s’ha extret anteriorment, sequencia. exonerate.gff és l’arxiu que conté només els exons i sequencia.nuc.fa és el fitxer de sortida que contindrà el cDNA en format FASTA.
Fastatranslate
Finalment, un cop ja s’ha obtingut el cDNA en format FASTA, s’ha executat el programa Fastatranslate per obtenir la seqüència d’aminoàcids:
$ fastatranslate -f sequencia.nuc.fa -F 1 > nomproteinapython.pep.fa
on sequencia.nuc.fa és la subseqüència que conté el cDNA en format FASTA, l’argument -F 1 s’utiliza per limitar el marc de lectura escollit, en aquest cas, és el primer dels que ofereix el programa, i per últim, nomproteinapython.pep.fa és el fitxer de sortida que contindrà la seqüència d’aminoàcids.
T-COFFEE (Tree-based Consistency Objective Function for alignment Evaluation) és un programa que permet realitzar un alineament global de la proteïna resultant obtinguda a partir del genoma de la pitó, amb la proteïna query utilitzada en cada cas. El resultat, per tant, permet avaluar l'homologia entre la query i la seqüència predita. El T-COFFEE s’ha executat amb la següent comanda:
$ t_coffee nomproteina.pep.fa nomproteinapython.pep.fa > tcoffeenomproteina.fa
on nomproteina.pep.fa és la query, nomproteinapython.pep.fa, la seqüència obtinguda del fastatranslate i tcoffeenomproteina.fa és el fitxer que conté l’alineament entre la seqüència predita i la query.
Per últim, s’ha realitzat la predicció d’elements SECIS (Selenocysteine Insertion Sequence) i una segona predicció de la selenoproteïna estudiada, a través del programa online Seblastian.
Els elements SECIS són elements essencials per a la traducció de la selenoproteïna i per tant, donaran suport a les prediccions que s’han realitzat.
Aquesta cerca s’ha fet amb les subseqüències (genomicsequenciasecis.fa) extretes del fastasubseq. A l’hora de predir els elements SECIS cal tenir en compte només la cadena (directa o reversa) que contengui el gen (se sap quan s'observa el resultat de l'Exonerate).