Our supervisor gave us a list of the 25 selenoproteins known in the humans. We choose three of them in order to analyze.
We have chosen 3 selenoproteins: selenoprotein I, selenoprotein N, selenoprotein O, which SecIS elements have been described previously.
All the following steps that we are going to describe have been applied to the three selenoproteins.
There are different databases, but we have chosen ensembl database because it recognizes the U of selenocysteines and distinguishes it from a stop codon.
First of all we used ensembl database in order to map our selenoproteins in the human genome. Ensembl develops a software system, on-line and free, which produces and maintains automatic annotation on metazoan genomes. We utilyze the option blast (Basic Local Alignment Search Tool ) / ssaha (Sequence Search and Alignment by Hashing Algorithm). It is used for sequence similarity searches, where ssaha is a very fast tool for matching and aligning DNA sequences.
This option of the ensembl permits us (thanks to blast and depending on query and subject libraries sequences) make use of different algorithms. We select tblastn option, used for a protein query versus a DNA library search.
This is necessary to obtain the DNA sequence from our original peptid selenoprotein sequence. To make this we paste our peptide sequence in fasta format, select peptide queries, and run them against the human DNA database. The program was configured selecting the smaller
e-value (0.0001), and with a high blosum (scoring matrix file) in order to obtain the best aligns and a faster running of the program.
With the results obtained, we select the ensembl transcript report. In this report we get all the information about the gene mapping. We can obtain the identifier from the gene, the genomic location (chromosome), strand, start/end of transcription, start/end of traduction, exons number and start/end of each exon. The results are also shown in a graph of whole sequence where we can select different options (exons, SNPs, codons, etc) to remark them to see them in an easier way.
Finding SNPs
Single Nucleotide Polymorphisms (SNPs) are DNA sequence variations that occur when a single nucleotide
(A,T,C,or G) in the genome sequence is changed. Most SNPs, actually about two of every three SNPs, involve the replacement of
cytosine (C) with thymine (T). SNPs occur every 100 to 300 bases along the human genome. SNPs are stable from an evolutionarily
standpoint --not changing much from generation to generation-- making them easier to follow in population studies.
Each person's genetic material contains a unique SNP pattern that is made up of many different genetic variations. Researchers have found that most SNPs are not responsible for a disease state. Instead, they serve as biological markers for pinpointing a disease on the human genome map, because they are usually located near a gene found to be associated with a certain disease. Occasionally, a SNP may actually cause a disease and, therefore, can be used to search for and isolate the disease-causing gene. In this project, we are looking for SNPs involved in diseases related with selenoprotein.
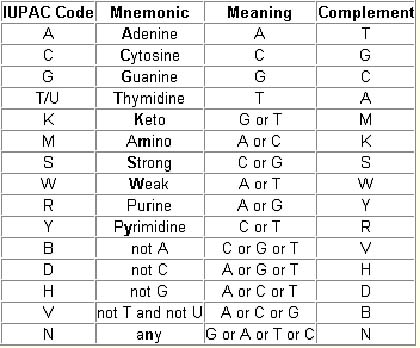
In the transcript report we choose the transcript cDNA sequence where we can select an option (codons/peptids/SNPs) that shows us the localization of all the SNPs in exons or UTR regions. This option allows us to see the importance of each SNP: if the polymorphism is a synonymous change or not thanks to the IUPAC ambiguity code and if the polymorphism affects a selenocysteine or the SecIS element.
Analyzing SecIS elements structure
In the ensembl transcript report there is the possibility of extracting transcript data in fasta file. We can choose it in the option �Export data: export transcript data in EMBL, GenBank or FASTA�. So we can select only the part of the sequence in which we are interested. We select, FASTA file and only the 3�UTR in the select sequence type, where there is the SecIS element.
We export this data to the SECISearch 2.19 program, which is a on-line server. This program is used to predict SecIS elements in the nucleotide sequence pasted. The program contains 3 modules, one to search sequence patterns, another one to evaluate the thermodynamic energy and just another one which permits seeing the predict structure.
We obtain the different pictures of the SecIS structures.
Analyzing SecIS elements
We use Blast 2.0 (Basic Local Alignment Search Tool), because it provides a method for rapid searching of nucleotide and protein databases. The BLAST algorithm detects local and global alignments.
First of all we keep the SecIS sequence and we use the Blast database to analyze these sequences.
Thanks to this program we make a search in the est_human database and we obtain a align with the ests sequences.
Ests sequences refers to expressed sequence tags. The ests are small pieces of DNA sequence that represent genes expressed in certain cells, tissues, or organs from different organisms and we use these tags to relate our sequences to some kind of human tissue for example in this database. With these aligns we can get important information about the similarity of sequences between our SecIS elements and the est_human sequences. Because of the characteristic expression of the est sequences we can relate our SecIS sequences to some tissue or disease.
We select the option nucleotide-nucleotid Blast (also known as Blastn), an algorithm used for simple nucleotides against nucleotide searches.
There we can paste our sequence (SecIS sequence). We choose the est_human database and in the option for advanced blasting we select Homo Sapiens to reduce the organisms range and make the search more specific and fast.
We made a program (findsnp.pl) based on perl programming and on the blastn output, with the objective to obtain a list containing: the different identifier of each sequence that contains a polymorphism, where are these SNPs (position) and which was the original nucleotide and which change has been produced. In the same way, we also made a second list, with the same program, in which we are able to see a recompilation of all the SNPs produced in all the sequences analyzed. So we know how many times one single SNP has been produced.
Thanks to this we obtain information on all the SNPs produced or related with our SecIS sequences.
Analyzing SecIS elements with SNPs
We also want to see the effect of the different SNPs affecting the SecIS elements.
For this reason we made a second program (Obtain_SecIS_sequence program.pl). In this case we now want to change our original SecIS sequence with each SNP that has been found.
We obtain as a result, all the SecIS sequences each one with a SNP. We have now a new list of SecIS sequences with one change in the nucleotide sequence corresponding to each SNP.
The objective now is to see if this SNPs affect the SecIS structure, so we have to use now again the SECIsearch program.
It is used as before with the same parameters, and we obtain the pictures of the different SecIS structures so we will be able to see if there is some important or specific change in the structure of the element.
Relation between SecIS sequence and diseases
We also want to see if there is a relation between the SecIS structure (and their SNPs) and some kind or specific disease.
Different ways were used to do this study:
- Using the information obtained from the Blast of Secis sequences against human est. In these results we can know the histological and clinical origin of the est sequence aligned with our sequence only checking the identifier and the brief of the sequence.
- Searching information in Pubmed Database(link) relating selenoproteins with diseases.
- Analysing Secis Structure with SeciSearch program and to observe if the structure differs from the normal one and can promote protein alterations.

