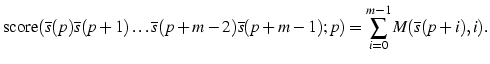
Amb el nostre programa aconseguim predir exons donada una seqüència. Utilitzem el següent esquema, on anem veient els mètodes seguits per realitzar cada pas:
Exons inicials (first): Aparellem els codons ATG amb els candidats a llocs de splicing donadors.
Exons interns (internal): Aparellem els candidats a llocs de splicing acceptors amb candidats a llocs de splicing donadors.
Exons terminals (terminal): Aparellem els candidats a llocs de splicing acceptors amb el codó stop de fi del interval.
Exons simples (single genes): Aparellem els codons ATG amb el codons stop de fi del interval.
Distingirem dos tipus de senyals de splicing: els donors i els acceptors. Els donors determinen l'extrem 5' dels introns, mentre que els acceptors determinen l'extrem 3' dels introns. Sota l'assumpció de que tractem amb senyals de splicing canònics, l'extrem 5' d'un intró comença amb el dinucleòtid GT, i l'extrem 3' acaba amb el dinucleòtid AG. Aquests dos dinucleòtids no només es troben en els extrems dels introns, sinó que també es poden trobar en qualsevol punt al llarg de la seqüència. Això fa que els senyals de splicing, ja siguin donors o acceptors, estiguin formats per una cadena de nucleòtids, en la qual es troba el punt de tall GT o AG, i el contingut de la qual ajuda a la maquinària de splicing a determinar el dinucleòtid GT, o AG, en aquella posició concreta, com a punt de tall.
Per tal d'establir on hi ha un senyal de splicing utilitzarem matrius de pesos (position weight matrices -PWMs): una pels senyals dels donors i una altra pels senyals dels acceptors. Aquestes matrius estaran enregistrades en fitxers de text en un format determinat (veure especificació més endavant), i el nostre programa ha de poder llegir-les d'aquests fitxers.
Considerem que tenim una matriu de pesos per identificar un senyal de splicing, donor o acceptor, que denotarem per M. Assumim que aquesta matriu està organitzada en 4 files, una per cada nucleòtid A,C,G,T i m columnes, una per cada posició del senyal, on les posicions aniran des de 0 fins a m-1. Ens referirem a una posició particular de la matriu indexant primer les files, i després les columnes. D'aquesta forma M(C,5) es refereix al pes associat al nucleòtid C quan el trobem a la sisena posició dins el senyal. Considerem que tenim una seqüència emmagatzemada en un vector s, tal que si la seqüència té n bases, el vector estarà indexat per les posicions 0 a n-1 i ens referirem a una base en particular com, per exemple, s(7) que especifica la vuitena base de la seqüència emmagatzemada al vector s. D'aquesta forma, la puntuació d'un possible senyal de splicing serà calculada sumant els pesos de la matriu que corresponen a un tros de seqüència que comença a la posició P i que estem examinant com a possible senyal de splicing:
Donada una seqüència amb n bases podrem puntuar un màxim de n-m+1 possibles senyals de splicing. Decidirem quins d'aquests senyals poden ser realment considerats com a tals d'acord amb un llindar en la puntuació, tal que només aquells trossos de seqüència que donen una puntuació superior al llindar seran considerats com a senyals de splicing.
Els exons que es tradueixen a proteïna solen mostrar un biaix cap a l'utilització de determinats codons, depenent de l'organisme al qual la seqüència d'ADN pertany. Aquest fet permet una interpretació en termes estadístics de la composició de codons d'un exó codificant. Aquesta interpretació consisteix en assumir que la distribució dels codons observada als introns és uniforme, mentre que la distribució dels codons observada als exons codificants divergeix notablement d'aquesta uniformitat.
Aquesta divergència es determina mitjançant la ponderació dels codons observats a la seqüència que analitzem, d'acord amb una taula d'ús dels codons a les regions codificants de l'organisme que estem analitzant. Donat un fragment de seqüència que es troba entre dos senyals de splicing, contra més codons observem que, d'acord amb la taula, apareixen freqüentment en regions codificants, direm que més gran serà el seu potencial codificant, i per tant serà més plausible que aquest fragment de seqüència correspongui a un exó.
Considerem una taula d'ús de codons que denotarem per T, i que estarà indexada pels codons de forma que T(ACT) es referirà a la proporció, en l'interval [0-1], d'ús del codó ACT en les regions codificants de l'organisme del qual s'ha derivat aquesta taula T. Assumim que tenim una seqüència emmagatzemada de la forma explicada a l'apartat anterior, i que volem calcular el biaix codificant d'una regió de q bases que va des de la posició p fins a la posició p+q-1 al vector s que emmagatzema la seqüència. Aquest biaix el calcularem com la raó de versemblança (o likelihood ratio en anglès) entre les proporcions esperades, que obtenim a partir de T, dels codons observats, i l'assumpció d'uniformitat per la qual la proporció esperada d'un codó qualsevol és 1/64:
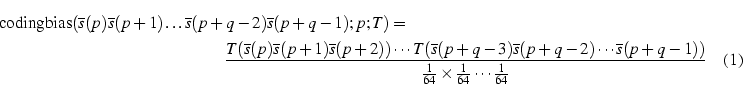
Els productes a l'expressió donaran com a resultat números extremadament petits, per tant haurem de treballar amb el logaritme de l'expressió anterior.
Entenem per pauta de lectura, o en anglès reading frame, d'un exó la posició de la primera base de l'exó, dins el seu codó, considerant 0 com la primera posició. Si la primera base de l'exó, correspon a la primera base del codó al qual pertany, direm que la pauta és 0, si aquesta primera base del exó és la segona base del seu codó, direm que la pauta és 1, i finalment, si la primera base de l'exó és la tercera base del seu codó, direm que la pauta és 2.
Quan predim un exó cal anotar la seva pauta de lectura, a més a més de les corresponents posicions d'inici i final. Com que aquesta pauta ve determinada per la divisió dels nucleòtids en codons, caldrà primer determinar quines divisions en codons són correctes. Una divisió correcta en codons és aquella en la que només podem trobar un codó de stop com a últim codó. Per tant caldrà trobar primer tots els possibles codons de stop i aixo ens determinarà el que es coneix com open reading frames (ORFs), o pautes obertes de lectura, que no són res més que parts de la seqüència que al dividir-les en codons, només podem trobar un codó de stop com a últim codó. Un cop tenim els ORFs, podrem anotar la pauta de lectura als exons que hem predit dins aquest ORF.
A més a més, respecte al l'exó d'inici, i a l'exó de final, del gen (coneguts en anglès com First i Terminal, respectivament) cal tenir en compte que el primer estarà determinat per un senyal de donor aparellat amb un codó d'inici (ATG), mentre que el segon estarà determinat per un senyal de acceptor aparellat amb un codó de stop (TAA, TGA, TAG). També busquem exons simples, corresponents als gens coneguts com a single gens, o gens d'un únic exó, que està delimitat entra un codó dinici i un codó stop.
El valor llindar dels exons predits és un cut-off que selecciona els exons que tenen una puntuació igual o superior al llindar seleccionat. Com més alt sigui el llindar, més restrictiva és la selecció de manera que predirem menys exons (ja que tindrem menys falsos positius); i a l'inrevés, a menor llindar, més exons predirem ja que augmentem la sensibilitat de la recerca.